
Alternative mRNA Processing of Innate Response Pathways in Respiratory Syncytial Virus (RSV) Infection



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus Preparation and Infection
2.3. Isoform-Selective RT-PCR
2.4. SMRT Sequencing
2.5. Short-Read RNA Sequencing
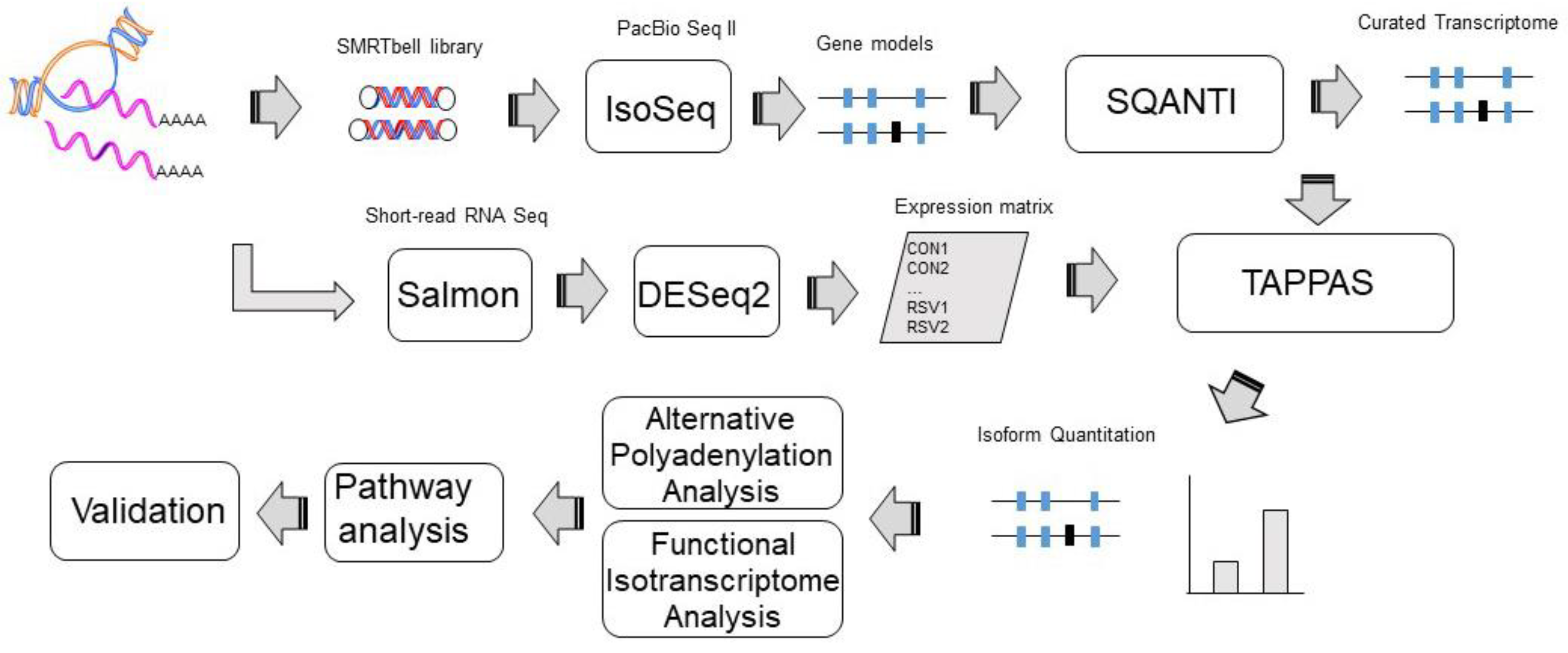
2.6. Data Analysis
3. Results
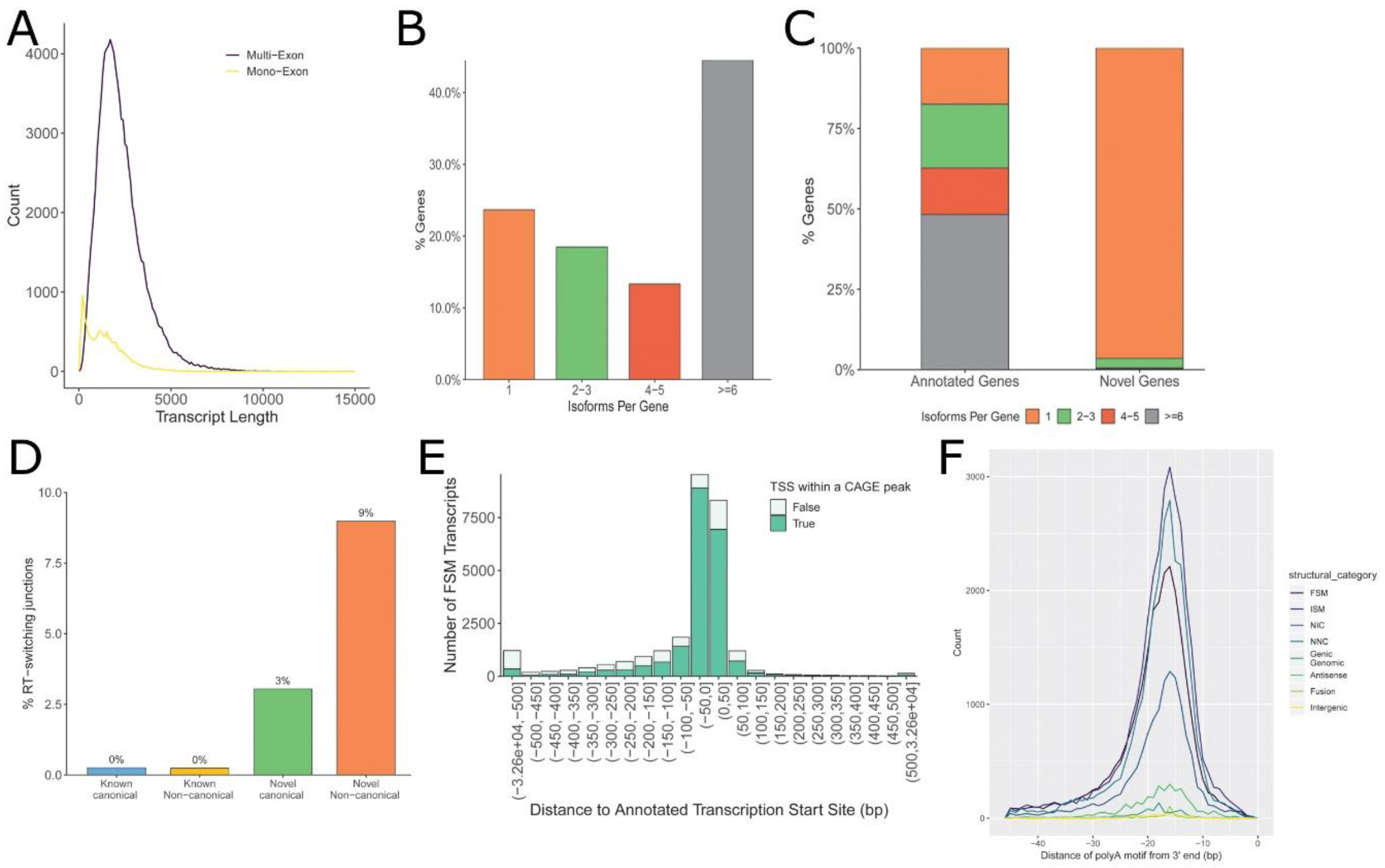
3.1. Annotation of hSAEC Transcriptome
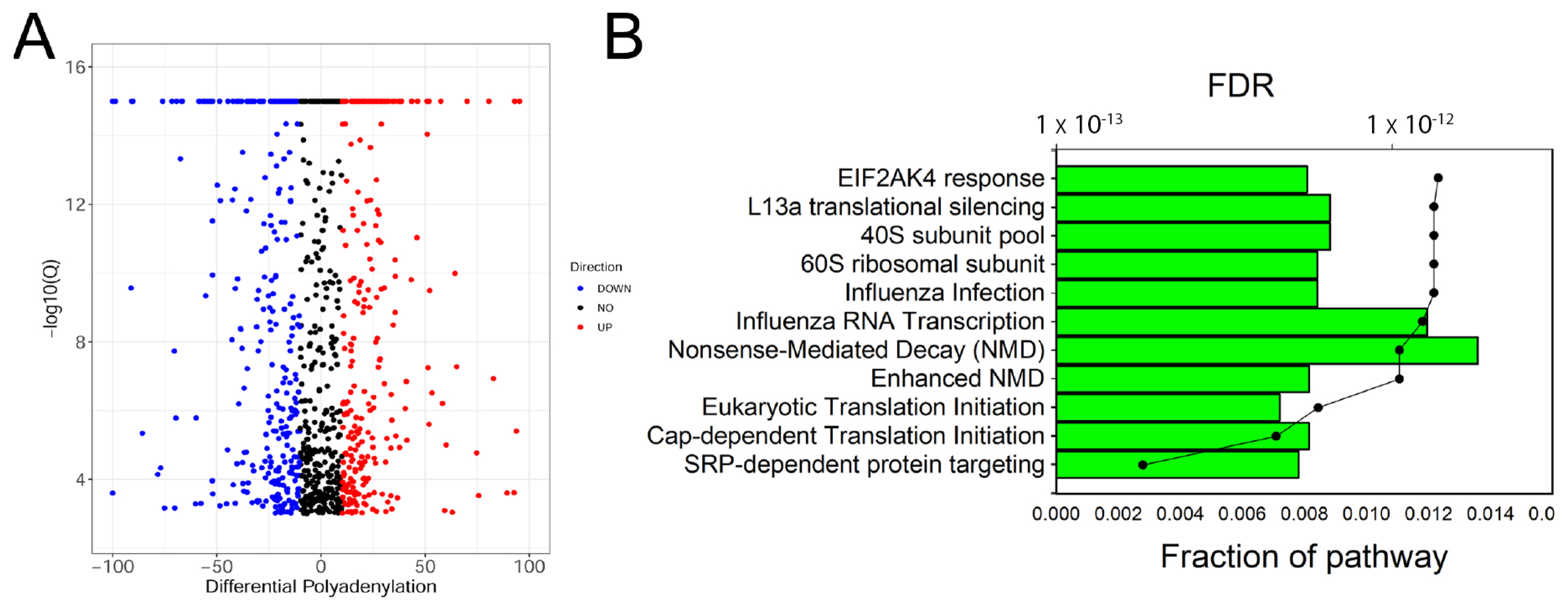
3.2. Alternative Polyadenylation (APA) Site Analysis
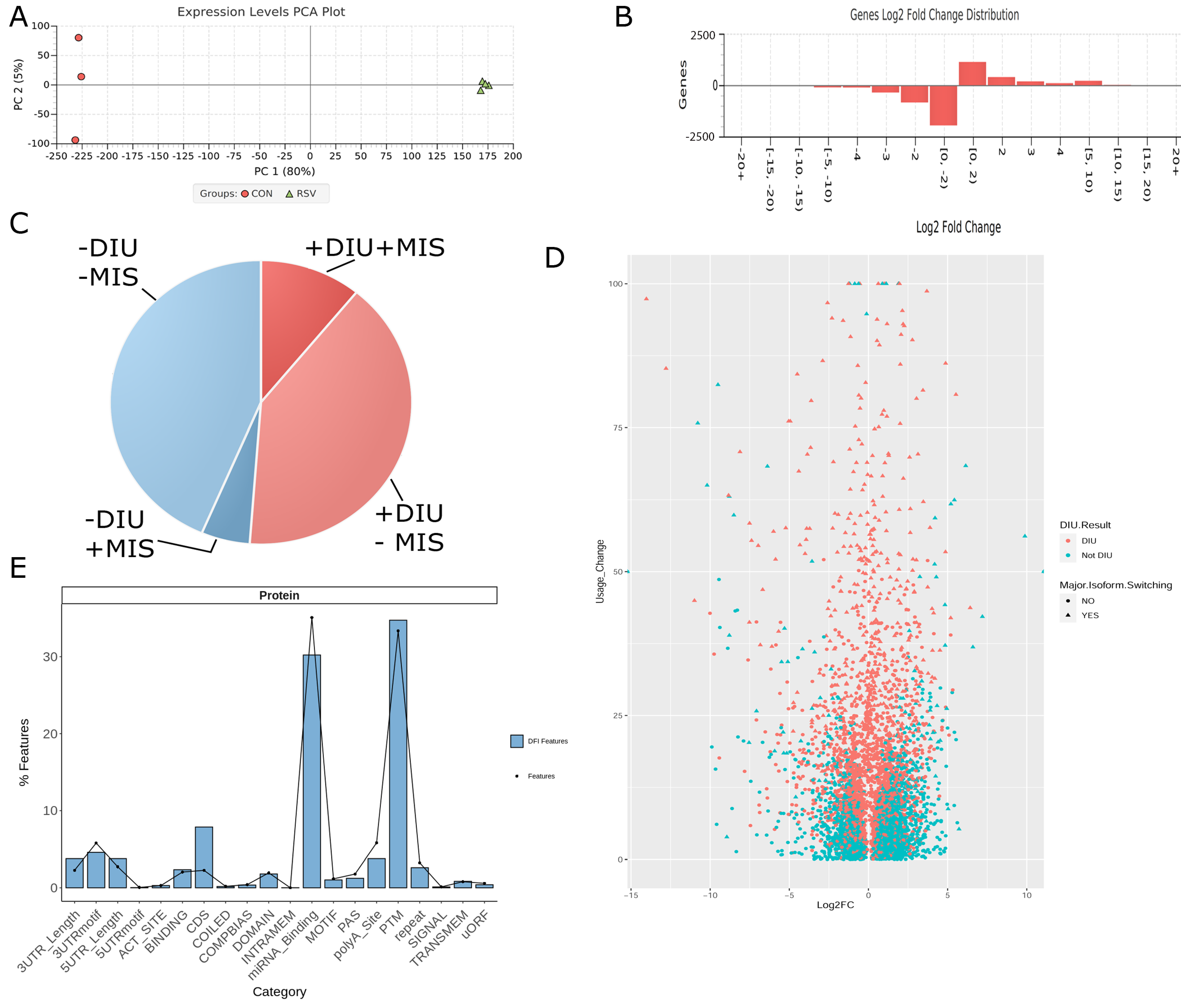
3.3. Differential Expression of Alternatively Spliced mRNA Isoforms Using Functional Iso-Transcriptomics (FIT) Analysis
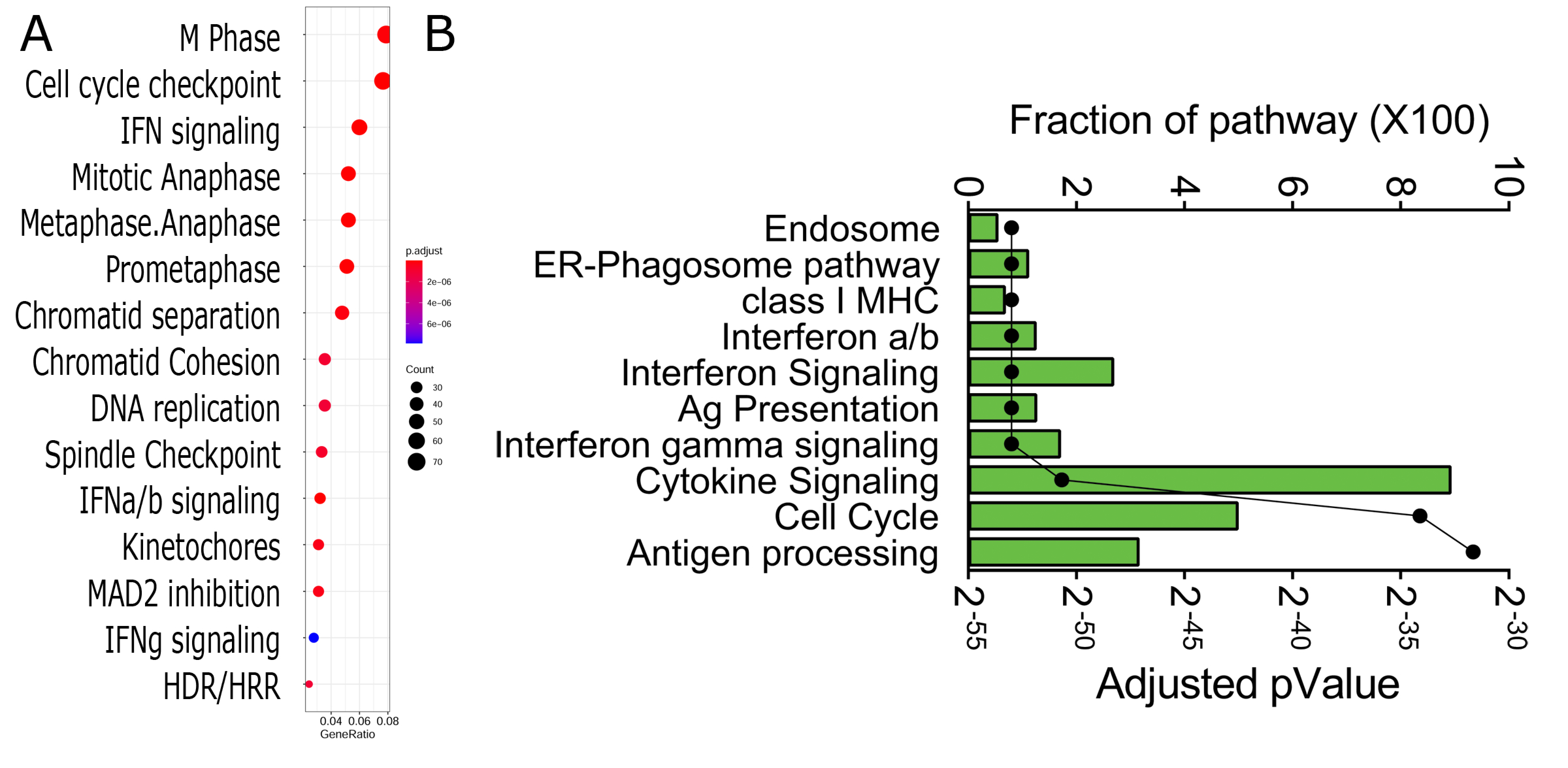
3.4. Functional Analysis
3.5. Network Analysis
3.6. Isoform Characterization
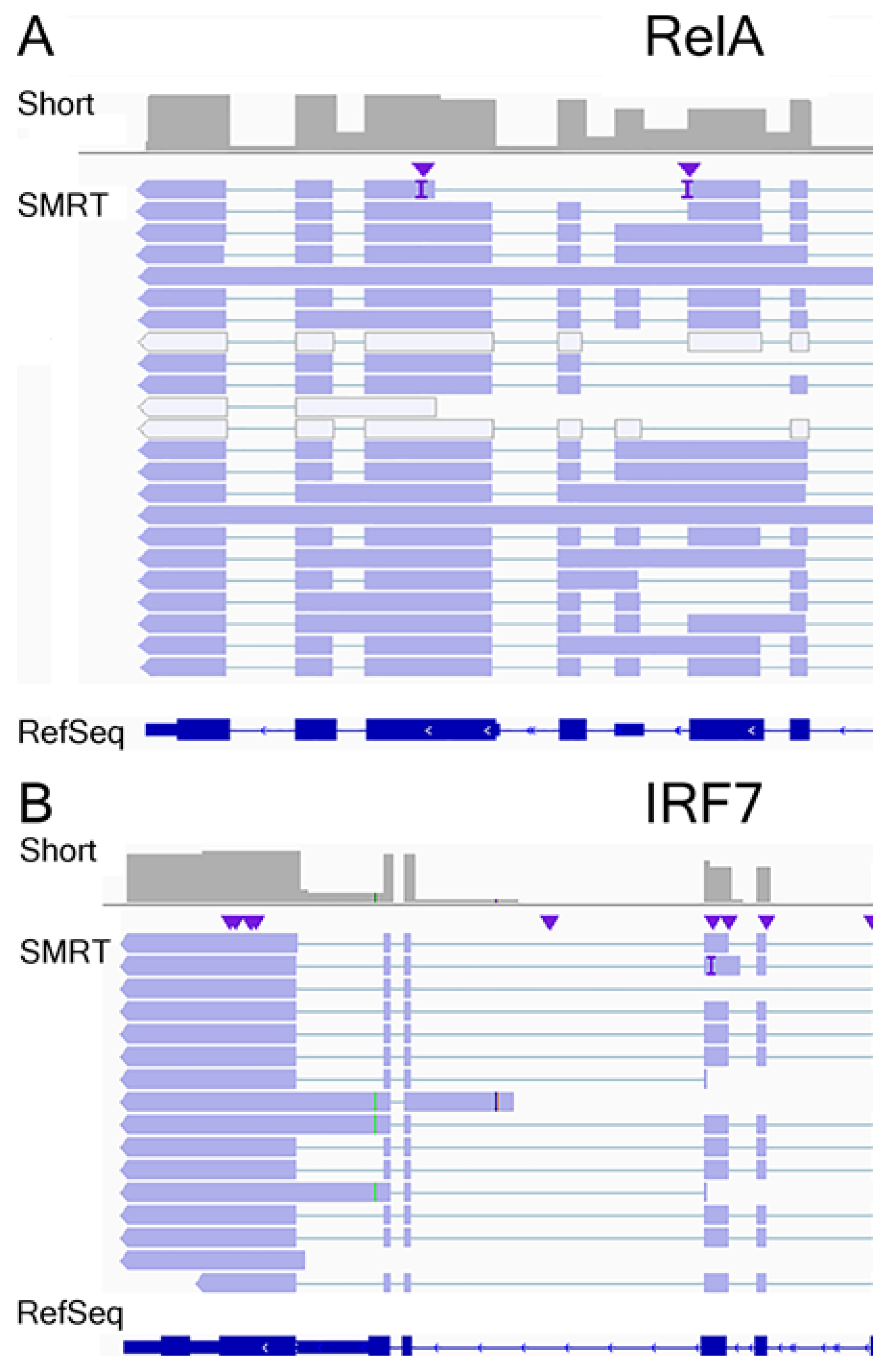
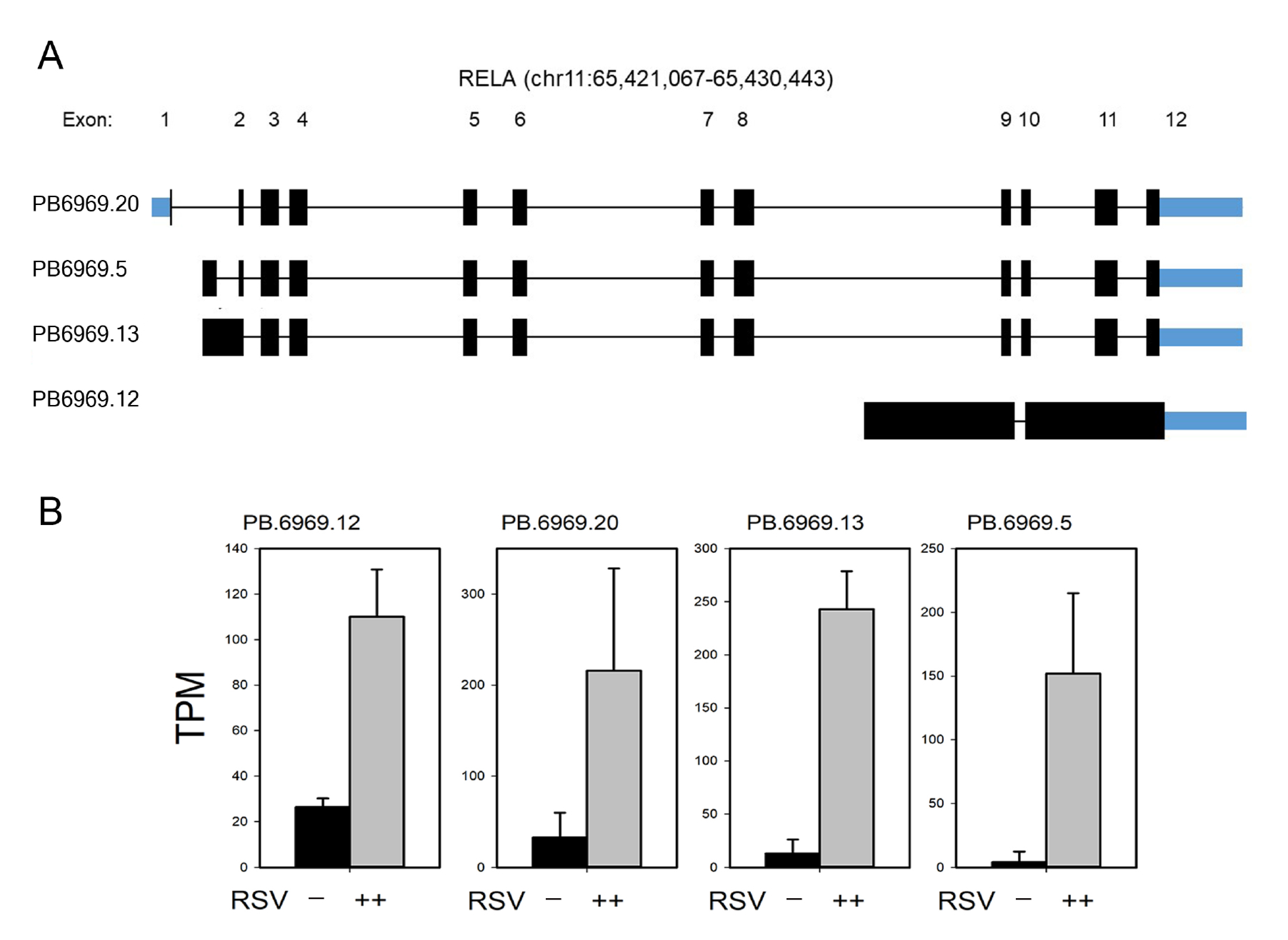
3.7. Annotation of RelA and IRF7 AS Isoforms
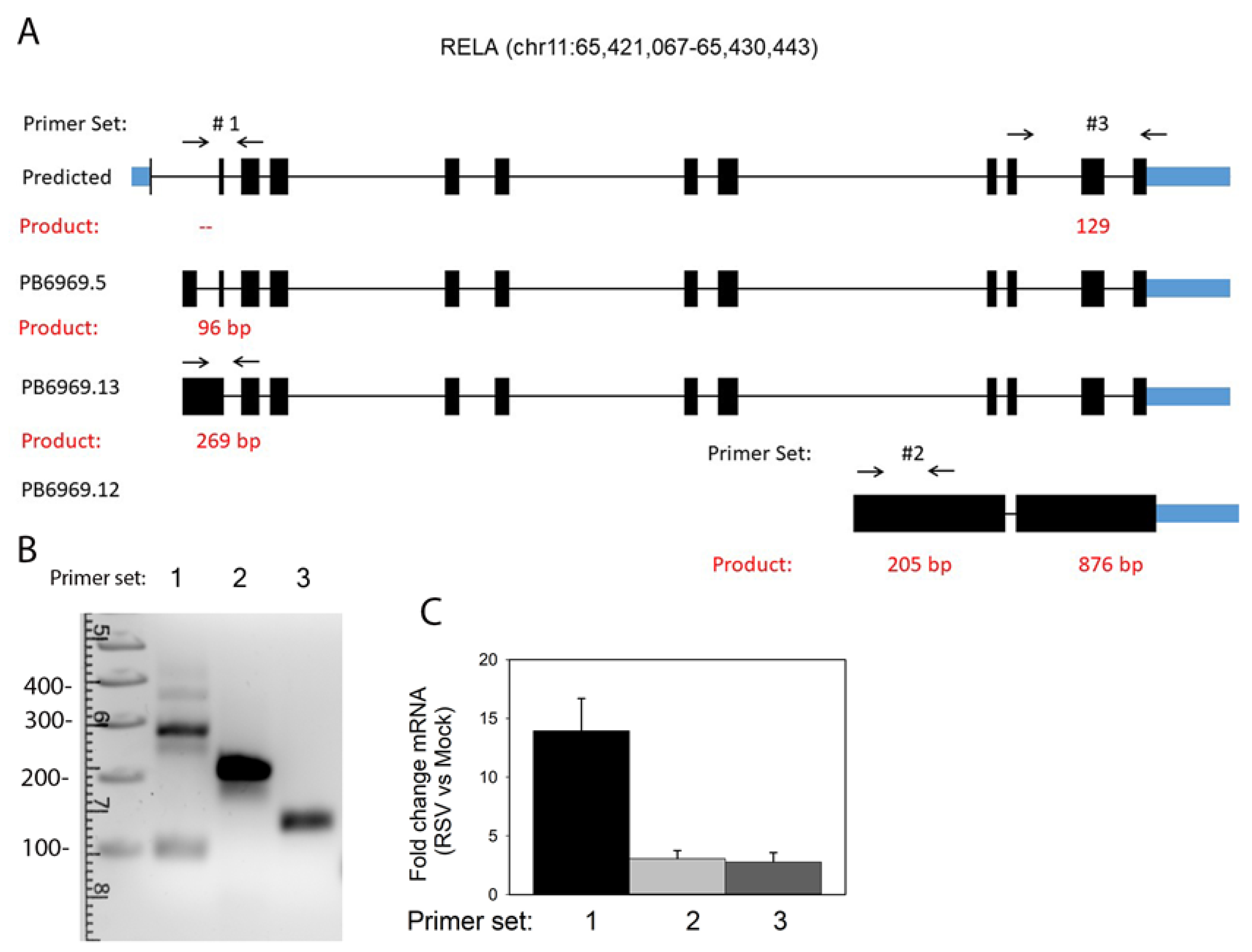
3.8. RelA Isoform Validation
3.9. IRF7 Isoform Validation
4. Discussion
4.1. SMRT Read Technology as a High-Resolution Method for Identifying AS and APA Regulation
4.2. mRNA Processing Mediates Cell Stress Responses
4.3. Mechanisms of How Transcriptional Initiation Is Coupled to Post-transcriptional mRNA Processing
4.4. AS of the RelA Transcription Factor
4.5. Inducible IRF7 mRNA Processing
4.6. Intron Retention (IR) in the IIR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pneumonia Etiology Research for Child Health Study Group. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: The PERCH multi-country case-control study. Lancet 2019, 394, 757–779. [CrossRef]
- Zhang, Y.; Luxon, B.A.; Casola, A.; Garofalo, R.P.; Jamaluddin, M.; Brasier, A.R. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J. Virol. 2001, 75, 9044–9058. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic Acid-Inducible Gene I Mediates Early Antiviral Response and Toll-Like Receptor 3 Expression in Respiratory Syncytial Virus-Infected Airway Epithelial Cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jamaluddin, M.; Zhang, Y.; Sun, H.; Ivanciuc, T.; Garofalo, R.P.; Brasier, A.R. Systematic Analysis of Cell-Type Differences in the Epithelial Secretome Reveals Insights into the Pathogenesis of Respiratory Syncytial Virus-Induced Lower Respiratory Tract Infections. J. Immunol. 2017, 198, 3345–3364. [Google Scholar] [CrossRef]
- Hosakote, Y.M.; Brasier, A.R.; Casola, A.; Garofalo, R.P.; Kurosky, A. Respiratory Syncytial Virus Infection Triggers Epithelial HMGB1 Release as a Damage-Associated Molecular Pattern Promoting a Monocytic Inflammatory Response. J. Virol. 2016, 90, 9618–9631. [Google Scholar] [CrossRef]
- Lay, M.K.; Bueno, S.M.; Galvez, N.; Riedel, C.A.; Kalergis, A.M. New insights on the viral and host factors contributing to the airway pathogenesis caused by the respiratory syncytial virus. Crit Rev. Microbiol 2016, 42, 800–812. [Google Scholar] [CrossRef]
- Bennett, B.L.; Garofalo, R.P.; Cron, S.G.; Hosakote, Y.M.; Atmar, R.L.; Macias, C.G.; Piedra, P.A. Immunopathogenesis of respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2007, 195, 1532–1540. [Google Scholar] [CrossRef]
- Jozwik, A.; Habibi, M.S.; Paras, A.; Zhu, J.; Guvenel, A.; Dhariwal, J.; Almond, M.; Wong, E.H.; Sykes, A.; Maybeno, M.; et al. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat. Commun. 2015, 6, 10224. [Google Scholar] [CrossRef]
- Garofalo, R.P.; Haeberle, H. Epithelial regulation of innate immunity to respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2000, 23, 581–585. [Google Scholar] [CrossRef]
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2018, 11, 21–34. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Lu, M.; Tian, B.; Li, K.; Garofalo, R.P.; Prusak, D.; Wood, T.G.; Brasier, A.R. Expression of an IKKgamma splice variant determines IRF3 and canonical NF-kappaB pathway utilization in ssRNA virus infection. PLoS ONE 2009, 4, e8079. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Czerkies, M.; Korwek, Z.; Prus, W.; Kochanczyk, M.; Jaruszewicz-Blonska, J.; Tudelska, K.; Blonski, S.; Kimmel, M.; Brasier, A.R.; Lipniacki, T. Cell fate in antiviral response arises in the crosstalk of IRF, NF-kappaB and JAK/STAT pathways. Nat. Commun. 2018, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhang, Y.; Luxon, B.A.; Garofalo, R.P.; Casola, A.; Sinha, M.; Brasier, A.R. Identification of NF-kappaB-dependent gene networks in respiratory syncytial virus-infected cells. J. Virol. 2002, 76, 6800–6814. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiao, D.; Mann, M.; Garofalo, R.P.; Brasier, A.R. Respiratory Syncytial Virus Infection Induces Chromatin Remodeling to Activate Growth Factor and Extracellular Matrix Secretion Pathways. Viruses 2020, 12, 804. [Google Scholar] [CrossRef]
- Zhang, Y.; Jamaluddin, M.; Wang, S.; Tian, B.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Ribavirin treatment up-regulates antiviral gene expression via the interferon-stimulated response element in respiratory syncytial virus-infected epithelial cells. J. Virol. 2003, 77, 5933–5947. [Google Scholar] [CrossRef]
- Tian, B.; Nowak, D.E.; Jamaluddin, M.; Wang, S.; Brasier, A.R. Identification of Direct Genomic Targets Downstream of the Nuclear Factor-κB Transcription Factor Mediating Tumor Necrosis Factor Signaling. J. Biol. Chem. 2005, 280, 17435–17448. [Google Scholar] [CrossRef]
- Tian, B.; Li, X.; Kalita, M.; Widen, S.G.; Yang, J.; Bhavnani, S.K.; Dang, B.; Kudlicki, A.; Sinha, M.; Kong, F.; et al. Analysis of the TGFbeta-induced program in primary airway epithelial cells shows essential role of NF-kappaB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genom. 2015, 16, 529. [Google Scholar] [CrossRef]
- Ramirez, R.D.; Sheridan, S.; Girard, L.; Sato, M.; Kim, Y.; Pollack, J.; Peyton, M.; Zou, Y.; Kurie, J.M.; Dimaio, J.M.; et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004, 64, 9027–9034. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Yang, J.; Zhao, Y.; Ivanciuc, T.; Sun, H.; Garofalo, R.P.; Brasier, A.R. Bromodomain Containing 4 (BRD4) Couples NFκB/RelA With Airway Inflammation And The IRF-RIG-I Amplification Loop In Respiratory Syncytial Virus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Zhang, J.; Jamaluddin, M.; Zhang, Y.; Widen, S.G.; Sun, H.; Brasier, A.R.; Zhao, Y. Type ii epithelial-mesenchymal transition upregulates protein n-glycosylation to maintain proteostasis and extracellular matrix production. J. Proteome. Res. 2019, 18, 3447–3460. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Yang, J.; Zhao, Y.; Ivanciuc, T.; Sun, H.; Garofalo, R.P.; Brasier, A.R. Bromodomain containing 4 (brd4) couples nfkb/rela with airway inflammation and the irf-rig-i amplification loop in respiratory syncytial virus infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Yang, J.; Zhao, Y.; Ivanciuc, T.; Sun, H.; Wakamiya, M.; Garofalo, R.P.; Brasier, A.R. Central Role of the NF-kappaB Pathway in the Scgb1a1-Expressing Epithelium in Mediating Respiratory Syncytial Virus-Induced Airway Inflammation. J. Virol. 2018, 92, e00441-00418. [Google Scholar] [CrossRef]
- Ueba, O. Respiratory syncytial virus: I. concentration and purification of the infectious virus. Acta Med. Okayama 1978, 32, 265–272. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef]
- Tardaguila, M.; de la Fuente, L.; Marti, C.; Pereira, C.; Pardo-Palacios, F.J.; Del Risco, H.; Ferrell, M.; Mellado, M.; Macchietto, M.; Verheggen, K.; et al. SQANTI: Extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 2018, 28, 396–411. [Google Scholar] [CrossRef]
- de la Fuente, L.; Arzalluz-Luque, Á.; Tardáguila, M.; del Risco, H.; Martí, C.; Tarazona, S.; Salguero, P.; Scott, R.; Lerma, A.; Alastrue-Agudo, A.; et al. tappAS: A comprehensive computational framework for the analysis of the functional impact of differential splicing. Genome Biol. 2020, 21, 119. [Google Scholar] [CrossRef] [PubMed]
- Cocquet, J.; Chong, A.; Zhang, G.; Veitia, R.A. Reverse transcriptase template switching and false alternative transcripts. Genomics 2006, 88, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Au, K.F.; Sebastiano, V.; Afshar, P.T.; Durruthy, J.D.; Lee, L.; Williams, B.A.; van Bakel, H.; Schadt, E.E.; Reijo-Pera, R.A.; Underwood, J.G.; et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, E4821–E4830. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, H.; Jahanbani, F.; Blauwkamp, T.; Moshrefi, A.; Jaeger, E.; Chen, F.; Harel, I.; Bustamante, C.D.; Rasmussen, M.; Snyder, M.P. Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events. Nat. Biotechnol. 2015, 33, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.; Peeples, M.; Hamilton, R. Effect of Respiratory Syncytial Virus infection of HeLa-cell macromolecular synthesis. J. Gen. Virol. 1977, 37, 53–63. [Google Scholar] [CrossRef]
- Odendall, C.; Kagan, J.C. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol. 2015, 12, 47–52. [Google Scholar] [CrossRef]
- Han, Y.; Whitney, G.; Donovan, J.; Korennykh, A. Innate immune messenger 2-5A tethers human RNase L into active high-order complexes. Cell Rep. 2012, 2, 902–913. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Kochs, G. Interferon-induced Mx proteins in antiviral host defense. Biochimie 2007, 89, 812–818. [Google Scholar] [CrossRef]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Reviews. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Furlanis, E.; Scheiffele, P. Regulation of Neuronal Differentiation, Function, and Plasticity by Alternative Splicing. Annu. Rev. Cell Dev. Biol. 2018, 34, 451–469. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cheng, Y.; Wu, W.; Liu, Y.; Wei, N.; Feng, X.; Xie, Z.; Feng, Y. SRSF10 regulates alternative splicing and is required for adipocyte differentiation. Mol. Cell. Biol. 2014, 34, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Reviews. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, J.A.; Whitworth, G.B.; Bergkessel, M.; Guthrie, C. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol. Cell 2007, 27, 928–937. [Google Scholar] [CrossRef]
- Gibbs, J.D.; Ornoff, D.M.; Igo, H.A.; Zeng, J.Y.; Imani, F. Cell cycle arrest by transforming growth factor beta1 enhances replication of respiratory syncytial virus in lung epithelial cells. J. Virol. 2009, 83, 12424–12431. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Wang, S.; Garofalo, R.P.; Elliott, T.; Casola, A.; Baron, S.; Brasier, A.R. Ifn-beta mediates coordinate expression of antigen-processing genes in rsv-infected pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol 2001, 280, L248–L257. [Google Scholar] [CrossRef]
- Smieja, J.; Jamaluddin, M.; Brasier, A.R.; Kimmel, M. Model-based analysis of interferon-beta induced signaling pathway. Bioinformatics 2008, 24, 2363–2369. [Google Scholar] [CrossRef]
- Anvar, S.Y.; Allard, G.; Tseng, E.; Sheynkman, G.M.; de Klerk, E.; Vermaat, M.; Yin, R.H.; Johansson, H.E.; Ariyurek, Y.; den Dunnen, J.T.; et al. Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. Genome Biol 2018, 19, 46. [Google Scholar] [CrossRef]
- Brasier, A.R.; Tian, B.; Jamaluddin, M.; Kalita, M.K.; Garofalo, R.P.; Lu, M. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J. Virol. 2011, 85, 11752–11769. [Google Scholar] [CrossRef]
- Brasier, A.R. RSV Reprograms the CDK9•BRD4 Chromatin Remodeling Complex to Couple Innate Inflammation to Airway Remodeling. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Kalita, M.; Li, X.; Jamaluddin, M.; Tian, B.; Edeh, C.B.; Wiktorowicz, J.E.; Kudlicki, A.; Brasier, A.R. Systematic Determination of Human Cyclin Dependent Kinase (CDK)-9 Interactome Identifies Novel Functions in RNA Splicing Mediated by the DDX5/17 RNA Helicases. Mol. Cell Proteom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Jantzi, P.D.; Esham, D.L.; Spratt, H.; Kurosky, A.; Casola, A.; Garofalo, R.P. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2011, 183, 1550–1560. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The NF-kappaB regulatory network. Cardiovasc Toxicol 2006, 6, 111–130. [Google Scholar] [CrossRef]
- Leeman, J.R.; Gilmore, T.D. Alternative splicing in the NF-kappaB signaling pathway. Gene 2008, 423, 97–107. [Google Scholar] [CrossRef]
- Narayanan, R.; Klement, J.F.; Ruben, S.M.; Higgins, K.A.; Rosen, C.A. Identification of a naturally occurring transforming variant of the p65 subunit of NF-kappa B. Science 1992, 256, 367–370. [Google Scholar] [CrossRef]
- Ruben, S.M.; Narayanan, R.; Klement, J.F.; Chen, C.H.; Rosen, C.A. Functional characterization of the NF-kappa B p65 transcriptional activator and an alternatively spliced derivative. Mol. Cell. Biol. 1992, 12, 444–454. [Google Scholar] [CrossRef]
- Zhang, L.; Pagano, J.S. Structure and function of IRF-7. J. Interferon Cytokine Res. 2002, 22, 95–101. [Google Scholar] [CrossRef]
- Frankiw, L.; Majumdar, D.; Burns, C.; Vlach, L.; Moradian, A.; Sweredoski, M.J.; Baltimore, D. BUD13 Promotes a Type I Interferon Response by Countering Intron Retention in Irf7. Mol. Cell 2019, 73, 803–814.e806. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Au, A.Y.; Ritchie, W.; Rasko, J.E. Intron retention in mRNA: No longer nonsense: Known and putative roles of intron retention in normal and disease biology. Bioessays 2016, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Baltimore, D. RNA splicing regulates the temporal order of TNF-induced gene expression. Proc. Natl. Acad. Sci. USA 2013, 110, 11934–11939. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene (Pair) | Forward Primer (5′–3′) | Reverse Primer | Target Transcript | Size (nt) |
|---|---|---|---|---|
| RelA 1 | ATTCCTTACCCCGTTTTCCCTC | AATGATCTCCACATAGGGGCCA | PB6969.5 | 96 |
| PB6969.13 | 269 | |||
| RelA-WT | None | |||
| RelA 2 | CATGTGGTCTTAGAGGGGCAG | GAGAAGTGGGACTTGCTCTCC | PB6969.12 | 205 |
| RelA-WT | None | |||
| RelA 3 | TCCTTTCAGCGGACCCAC | TTGATGGTGCTCAGGGATGAC | PB6969.12 | 876 |
| RelA-WT | 129 | |||
| IRF7 4 | GCTGCTTACGTTCACCCTGAC | CTTGGAGTCCAGCATGTGTGTG | PB6624.7 | 82 |
| IRF7-WT | None | |||
| IRF7 5 | AAGAAGGGCTTCCCCTGACT | CTCTACTGCCCACCCGTACA | PB6624.7 | 216 |
| IRF7-WT | 80 | |||
| IRF7 6 | GAGGCCCGCACCTGTTT | TTGGGGAGGGTGACAGGTA | PB123733 | 123 |
| IRF7 | 637/724 | |||
| IRF7 7 | AGCCCTTACCTCCCCTGTTA | GCTGATCTCTCCAAGGAGCC | PB6624.10/PB123733 | 130 |
| IRF7-WT | 215 | |||
| IRF7-8 | CTCGGAACTGTGACACCCCC | AGCCCAGGTAGATGGTATAGCG | PB6624.10 | 199 |
| IRF7-WT | 111 |
| Category | Isoform (No.) |
|---|---|
| FSM | 28,561 |
| ISM | 35,495 |
| NIC | 14,972 |
| NNC | 28,166 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Mann, M.; Qiao, D.; Brasier, A.R. Alternative mRNA Processing of Innate Response Pathways in Respiratory Syncytial Virus (RSV) Infection. Viruses 2021, 13, 218. https://doi.org/10.3390/v13020218
Xu X, Mann M, Qiao D, Brasier AR. Alternative mRNA Processing of Innate Response Pathways in Respiratory Syncytial Virus (RSV) Infection. Viruses. 2021; 13(2):218. https://doi.org/10.3390/v13020218
Chicago/Turabian StyleXu, Xiaofang, Morgan Mann, Dianhua Qiao, and Allan R. Brasier. 2021. "Alternative mRNA Processing of Innate Response Pathways in Respiratory Syncytial Virus (RSV) Infection" Viruses 13, no. 2: 218. https://doi.org/10.3390/v13020218
APA StyleXu, X., Mann, M., Qiao, D., & Brasier, A. R. (2021). Alternative mRNA Processing of Innate Response Pathways in Respiratory Syncytial Virus (RSV) Infection. Viruses, 13(2), 218. https://doi.org/10.3390/v13020218