
Potential Dual Role of West Nile Virus NS2B in Orchestrating NS3 Enzymatic Activity in Viral Replication

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus
2.2. Plasmid Construction
2.3. Transient Transfections and Protein Expression
2.4. Antibodies
2.5. Indirect Immunofluorescence Assay and Image Acquisition
2.6. Quantitation of Colocalization
2.7. Cell Lysis
2.8. Subcellular Fractionation
2.9. Western Blot
3. Results
3.1. NS3 and NS2B Localize to the ER in WNV-Infected Cells
3.2. WNV NS3 Is Distributed throughout the Cytoplasm, While NS2B Colocalizes with the ER in Transfected Cells
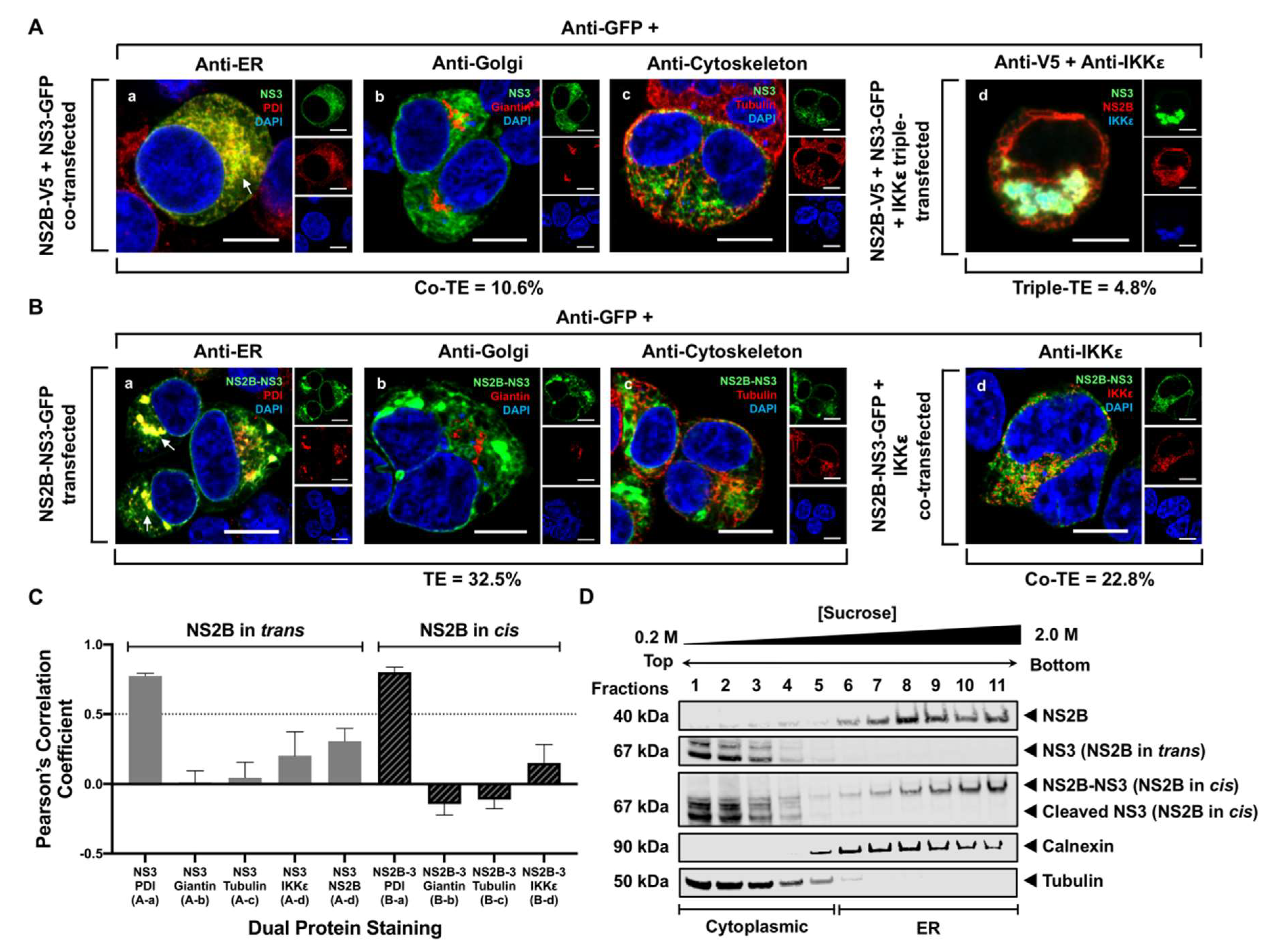
3.3. NS3 Localizes to the ER When NS2B Is Provided in Trans and in Cis
3.4. NS3 Is Proteolytically Active When NS2B Provided in Trans
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heinz, F.X.; Collett, M.S.; Purcell, R.H.; Gould, E.A.; Howard, C.R.; Houghton, M.; Moormann, R.J.; Rice, C.M.; Tiehl, H.J. Flaviviridae. In Virus taxonomy: Classification and nomenclature of viruses. 7th Report of the International Committee for the Taxonomy of Viruses; van Regenmortel, M.H.V., Fauquet, C.M., Bishop, D.H.L., Carstens, E.B., Estes, M.K., Lemon, S.M., Maniloff, J., Mayo, M.A., McGeoch, D.J., Pringle, C.R., et al., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 869–878. [Google Scholar]
- Sejvar, J.J.; Haddad, M.B.; Tierney, B.C.; Campbell, G.L.; Marfin, A.A.; Van Gerpen, J.A.; Fleischauer, A.; Leis, A.A.; Stokic, D.S.; Petersen, L.R. Neurologic manifestations and outcome of West Nile virus infection. JAMA 2003, 290, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Nowak, T.; Farber, P.M.; Wengler, G.; Wengler, G. Analyses of the terminal sequences of West Nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology 1989, 169, 365–376. [Google Scholar] [CrossRef]
- Castle, E.; Leidner, U.; Nowak, T.; Wengler, G.; Wengler, G. Primary structure of the West Nile flavivirus genome region coding for all nonstructural proteins. Virology 1986, 149, 10–26. [Google Scholar] [CrossRef]
- Chu, J.J.H.; Rajamanonmani, R.; Li, J.; Bhuvanakantham, R.; Lescar, J.; Ng, M.L. Inhibition of West Nile virus entry by using a recombinant domain III from the envelope glycoprotein. J. Gen. Virol. 2005, 86, 405–412. [Google Scholar] [CrossRef] [PubMed]
- VanBlargan, L.A.; Davis, K.A.; Dowd, K.A.; Akey, D.L.; Smith, J.L.; Pierson, T.C. Context-Dependent Cleavage of the Capsid Protein by the West Nile Virus Protease Modulates the Efficiency of Virus Assembly. J. Virol. 2015, 89, 8632–8642. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Westaway, E.G. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: Colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar] [CrossRef]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Falgout, B.; Chanock, R.; Lai, C.J. Proper processing of dengue virus nonstructural glycoprotein NS1 requires the N-terminal hydrophobic signal sequence and the downstream nonstructural protein NS2a. J. Virol. 1989, 63, 1852–1860. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J. Virol. 1997, 71, 9608–9617. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Gayen, S.; Kang, C.; Yuan, Z.; Shi, P.Y. Membrane topology and function of dengue virus NS2A protein. J. Virol. 2013, 87, 4609–4622. [Google Scholar] [CrossRef] [PubMed]
- Clum, S.; Ebner, K.E.; Padmanabhan, R. Cotranslational membrane insertion of the serine proteinase precursor NS2B-NS3(Pro) of dengue virus type 2 is required for efficient in vitro processing and is mediated through the hydrophobic regions of NS2B. J. Biol. Chem. 1997, 272, 30715–30723. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Khromykh, A.A.; Jones, M.K.; Westaway, E.G. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 1998, 245, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular localization and membrane topology of the Dengue virus type 2 Non-structural protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Zhang, L.; Ramachandra, M.; Kusukawa, J.; Ebner, K.E.; Padmanabhan, R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 1995, 270, 19100–19106. [Google Scholar] [CrossRef]
- Li, H.; Clum, S.; You, S.; Ebner, K.E.; Padmanabhan, R. The serine protease and RNA-stimulated nucleoside triphosphatase and RNA helicase functional domains of dengue virus type 2 NS3 converge within a region of 20 amino acids. J. Virol. 1999, 73, 3108–3116. [Google Scholar] [CrossRef]
- Egloff, M.P.; Benarroch, D.; Selisko, B.; Romette, J.L.; Canard, B. An RNA cap (nucleoside-2’-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar] [CrossRef]
- Nomaguchi, M.; Teramoto, T.; Yu, L.; Markoff, L.; Padmanabhan, R. Requirements for West Nile virus (-)- and (+)-strand subgenomic RNA synthesis in vitro by the viral RNA-dependent RNA polymerase expressed in Escherichia coli. J. Biol. Chem. 2004, 279, 12141–12151. [Google Scholar] [CrossRef]
- Wengler, G.; Wengler, G. The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology 1991, 184, 707–715. [Google Scholar] [CrossRef]
- Falgout, B.; Pethel, M.; Zhang, Y.M.; Lai, C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991, 65, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.K.; Kuhn, R.J.; Smith, J.L. Functional characterization of cis and trans activity of the Flavivirus NS2B-NS3 protease. J. Biol. Chem. 2007, 282, 12883–12892. [Google Scholar] [CrossRef] [PubMed]
- Chernov, A.V.; Shiryaev, S.A.; Aleshin, A.E.; Ratnikov, B.I.; Smith, J.W.; Liddington, R.C.; Strongin, A.Y. The two-component NS2B-NS3 proteinase represses DNA unwinding activity of the West Nile virus NS3 helicase. J. Biol. Chem. 2008, 283, 17270–17278. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.M.; Thiel, C.L.; Rice, H.-J.; Flaviviridae, C.M. Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer/Lippencott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 712–746. [Google Scholar]
- Klema, V.J.; Padmanabhan, R.; Choi, K.H. Flaviviral Replication Complex: Coordination between RNA Synthesis and 5’-RNA Capping. Viruses 2015, 7, 4640–4656. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Huang, Z.S.; Chiang, P.L.; Chen, C.T.; Wu, H.N. Analysis of the nucleoside triphosphatase, RNA triphosphatase, and unwinding activities of the helicase domain of dengue virus NS3 protein. FEBS Lett. 2009, 583, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Q.; Wong, Y.L.; Liew, L.S.; Kang, C. Membrane topology of NS2B of dengue virus revealed by NMR spectroscopy. Biochim. Biophys. Acta 2015, 1848, 2244–2252. [Google Scholar] [CrossRef]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Chernov, A.V.; Aleshin, A.E.; Shiryaeva, T.N.; Strongin, A.Y. NS4A regulates the ATPase activity of the NS3 helicase: A novel cofactor role of the non-structural protein NS4A from West Nile virus. J. Gen. Virol. 2009, 90, 2081–2085. [Google Scholar] [CrossRef]
- Umareddy, I.; Chao, A.; Sampath, A.; Gu, F.; Vasudevan, S.G. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 2006, 87, 2605–2614. [Google Scholar] [CrossRef]
- Yu, L.; Takeda, K.; Markoff, L. Protein-protein interactions among West Nile non-structural proteins and transmembrane complex formation in mammalian cells. Virology 2013, 446, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Falgout, B.; Miller, R.H.; Lai, C.J. Deletion analysis of dengue virus type 4 nonstructural protein NS2B: Identification of a domain required for NS2B-NS3 protease activity. J. Virol. 1993, 67, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Su, X.C.; Ozawa, K.; Qi, R.; Vasudevan, S.G.; Lim, S.P.; Otting, G. NMR analysis of the dynamic exchange of the NS2B cofactor between open and closed conformations of the West Nile virus NS2B-NS3 protease. PLoS Negl. Trop. Dis. 2009, 3, e561. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Grakoui, A.; Rice, C.M. Processing of the yellow fever virus nonstructural polyprotein: A catalytically active NS3 proteinase domain and NS2B are required for cleavages at dibasic sites. J. Virol. 1991, 65, 6042–6050. [Google Scholar] [CrossRef]
- Kaufusi, P.H.; Tseng, A.C.; Kelley, J.F.; Nerurkar, V.R. Selective Reactivity of Anti-Japanese Encephalitis Virus NS4B Antibody Towards Different Flaviviruses. Viruses 2020, 12, 212. [Google Scholar] [CrossRef]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef]
- Kaufusi, P.H.; Tseng, A.; Nerurkar, V.R. Functional Analysis of West Nile Virus Proteins in Human Cells. Methods Mol. Biol. 2016, 1435, 45–60. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 1, pp. 1.10–17.79. [Google Scholar]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]
- Kaufusi, P.H.; Kelley, J.F.; Yanagihara, R.; Nerurkar, V.R. Induction of endoplasmic reticulum-derived replication-competent membrane structures by West Nile virus non-structural protein 4B. PLoS ONE 2014, 9, e84040. [Google Scholar] [CrossRef] [PubMed]
- Assenberg, R.; Mastrangelo, E.; Walter, T.S.; Verma, A.; Milani, M.; Owens, R.J.; Stuart, D.I.; Grimes, J.M.; Mancini, E.J. Crystal structure of a novel conformational state of the flavivirus NS3 protein: Implications for polyprotein processing and viral replication. J. Virol. 2009, 83, 12895–12906. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Y.; Saw, W.G.; Zhao, Y.; Chan, K.W.; Singh, D.; Chong, Y.; Forwood, J.K.; Ooi, E.E.; Gruber, G.; Lescar, J.; et al. The C-terminal 50 amino acid residues of dengue NS3 protein are important for NS3-NS5 interaction and viral replication. J. Biol. Chem. 2015, 290, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Lee, L.T.; Wang, Q.Y.; Xie, X.; Lu, S.; Yau, Y.H.; Yuan, Z.; Geifman Shochat, S.; Kang, C.; Lescar, J.; et al. Mapping the Interactions between the NS4B and NS3 proteins of dengue virus. J. Virol. 2015, 89, 3471–3483. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.J.; Ng, M.M.; Chow, V.T. The non-structural 3 (NS3) protein of dengue virus type 2 interacts with human nuclear receptor binding protein and is associated with alterations in membrane structure. Virus Res. 2004, 102, 151–163. [Google Scholar] [CrossRef]
- Uchil, P.D.; Kumar, A.V.; Satchidanandam, V. Nuclear localization of flavivirus RNA synthesis in infected cells. J. Virol. 2006, 80, 5451–5464. [Google Scholar] [CrossRef]
- Anglero-Rodriguez, Y.I.; Pantoja, P.; Sariol, C.A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IkappaB kinase epsilon interaction. Clin. Vaccine Immunol. 2014, 21, 29–38. [Google Scholar] [CrossRef]
- Pambudi, S.; Kawashita, N.; Phanthanawiboon, S.; Omokoko, M.D.; Masrinoul, P.; Yamashita, A.; Limkittikul, K.; Yasunaga, T.; Takagi, T.; Ikuta, K.; et al. A small compound targeting the interaction between nonstructural proteins 2B and 3 inhibits dengue virus replication. Biochem. Biophys. Res. Commun. 2013, 440, 393–398. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WNV Gene Constructs | Primer | Nucleotide Position * | Primer Sequence (5’ → 3’) |
|---|---|---|---|
| NS2B | forward | 4219–4234 | accaccatg a GGATGGCCCGCAACTG |
| reverse | 4686–4611 | TCTCTTTGTGTATTGGAGAGTTATCC | |
| NS3 | forward | 4612–4631 | accaccatg a GGAGGCGTGTTGTGGGACAC |
| reverse | 6444–6468 | GACGTTTTCCCGAGGCGAAGTCCTTG | |
| NS2B-NS3 | forward | 4219–4234 | accaccatg a GGATGGCCCGCAACTG |
| reverse | 6444–6468 | GACGTTTTCCCGAGGCGAAGTCCTTG | |
| NS2B-NS3-NS4A | forward | 4219–4234 | accaccatg a GGATGGCCCGCAACTG |
| reverse | 6824–6846 | GACGTTGCTTCTCTGGCTCAGGAA | |
| NS4B-NS5 | forward b | 6865–6882 | accaccatg a gct c CTAGCCGTGTTCCTGATT |
| reverse | 10370–10395 | GCAGTACTGTGTCCTCAACCAAAGTTG | |
| NS5 | forward | 7681–7698 | accaccatg a GTGGGGCAAAAGGACGC |
| reverse | 10370–10395 | GCAGTACTGTGTCCTCAACCAAAGTTG |
| Protein | Primary Antibody | Catalog Number | IF | WB | Secondary Antibody | Catalog Number | IF | WB |
|---|---|---|---|---|---|---|---|---|
| Dilutions | Dilutions | |||||||
| WNV NS2B | rabbit polyclonal | GTX132060 a | 1:100 | anti-rabbit IgG AF 488 | 1:1000 | A11008 b | ||
| 1:100 | anti-rabbit IgG AF 555 | A21428 b | 1:1000 | |||||
| 1:3000 | anti-rabbit IRDye 800CW | 926-32211 g | 1:10,000 | |||||
| WNV NS3 | rabbit polyclonal | GTX131955 a | 1:100 | anti-rabbit IgG AF 488 | A11008 b | 1:1000 | ||
| 1:100 | anti-rabbit IgG AF 555 | A21428 b | 1:1000 | |||||
| 1:3000 | anti-rabbit IRDye 800CW | 926-32211 g | 1:10,000 | |||||
| Calnexin | rabbit polyclonal | C4731 c | 1:100 | anti-rabbit IgG AF 488 | A11008 b | 1:1000 | ||
| 1:2000 | anti-rabbit IRDye 800CW | 926-32211 g | 1:10,000 | |||||
| Protein Disulfide Isomerase (PDI) | mouse monoclonal | MA3-019 b | 1:100 | anti-mouse IgG AF 555 | A21422 b | 1:1000 | ||
| Giantin | rabbit polyclonal | ab80864 d | 1:100 | anti-rabbit IgG AF 555 | A21428 b | 1:1000 | ||
| Golgi matrix 130 (GM130) | mouse monoclonal | 610822 e | 1:100 | anti-mouse IgG AF 555 | A21422 b | 1:1000 | ||
| Tubulin β | rabbit polyclonal | RB9249 b | 1:100 | anti-rabbit IgG AF 555 | A21428 b | 1:1000 | ||
| mouse monoclonal | sc-5274 f | 1:1000 | anti-mouse IRDye 680RD | 926-68170 g | 1:10,000 | |||
| IκB kinase subunit ε (IKKε) | rabbit polyclonal | ab7891 d | 1:100 | anti-rabbit IgG AF 555 | A21428 b | 1:1000 | ||
| 1:100 | anti-rabbit IgG AF 488 | A11008 b | 1:1000 | |||||
| 1:100 | anti-rabbit IgG Pacific Blue | P10994 b | 1:500 | |||||
| β-actin | mouse monoclonal | A5316 c | 1:2000 | anti-mouse IRDye 680RD | 926-68170 g | 1:10,000 | ||
| Green fluorescent protein (GFP) | rabbit polyclonal | G10362 b | 1:100 | anti-rabbit IgG AF 488 | A11008 b | 1:1000 | ||
| 1:4000 | anti-rabbit IRDye 800CW | 926-32211 g | 1:10,000 | |||||
| V5/His epitope | mouse monoclonal | R960-25 b | 1:100 | anti-mouse IgG AF 555 | A21422 b | 1:1000 | ||
| 1:2000 | anti-mouse IRDye 680RD | 926-68170 g | 1:10,000 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, A.C.; Nerurkar, V.R.; Neupane, K.R.; Kae, H.; Kaufusi, P.H. Potential Dual Role of West Nile Virus NS2B in Orchestrating NS3 Enzymatic Activity in Viral Replication. Viruses 2021, 13, 216. https://doi.org/10.3390/v13020216
Tseng AC, Nerurkar VR, Neupane KR, Kae H, Kaufusi PH. Potential Dual Role of West Nile Virus NS2B in Orchestrating NS3 Enzymatic Activity in Viral Replication. Viruses. 2021; 13(2):216. https://doi.org/10.3390/v13020216
Chicago/Turabian StyleTseng, Alanna C., Vivek R. Nerurkar, Kabi R. Neupane, Helmut Kae, and Pakieli H. Kaufusi. 2021. "Potential Dual Role of West Nile Virus NS2B in Orchestrating NS3 Enzymatic Activity in Viral Replication" Viruses 13, no. 2: 216. https://doi.org/10.3390/v13020216
APA StyleTseng, A. C., Nerurkar, V. R., Neupane, K. R., Kae, H., & Kaufusi, P. H. (2021). Potential Dual Role of West Nile Virus NS2B in Orchestrating NS3 Enzymatic Activity in Viral Replication. Viruses, 13(2), 216. https://doi.org/10.3390/v13020216