
Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188

 ,
,
Abstract
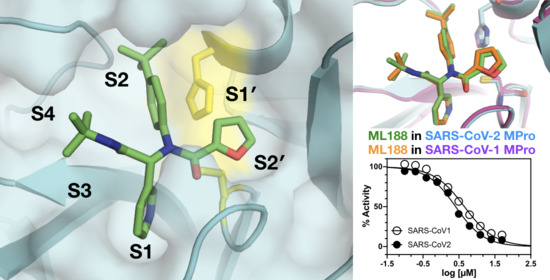
{kind=link}
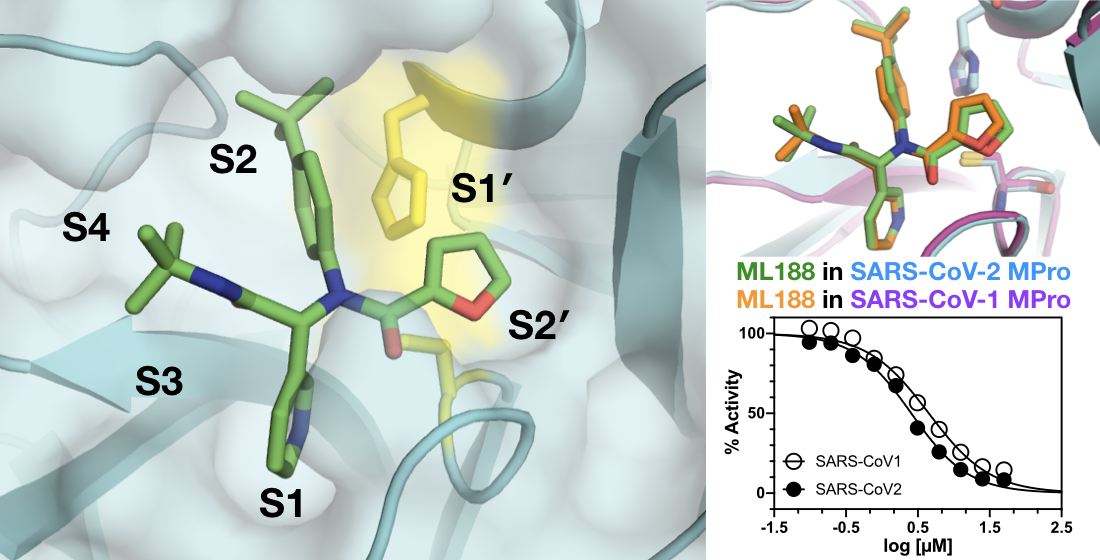
{kind=link}
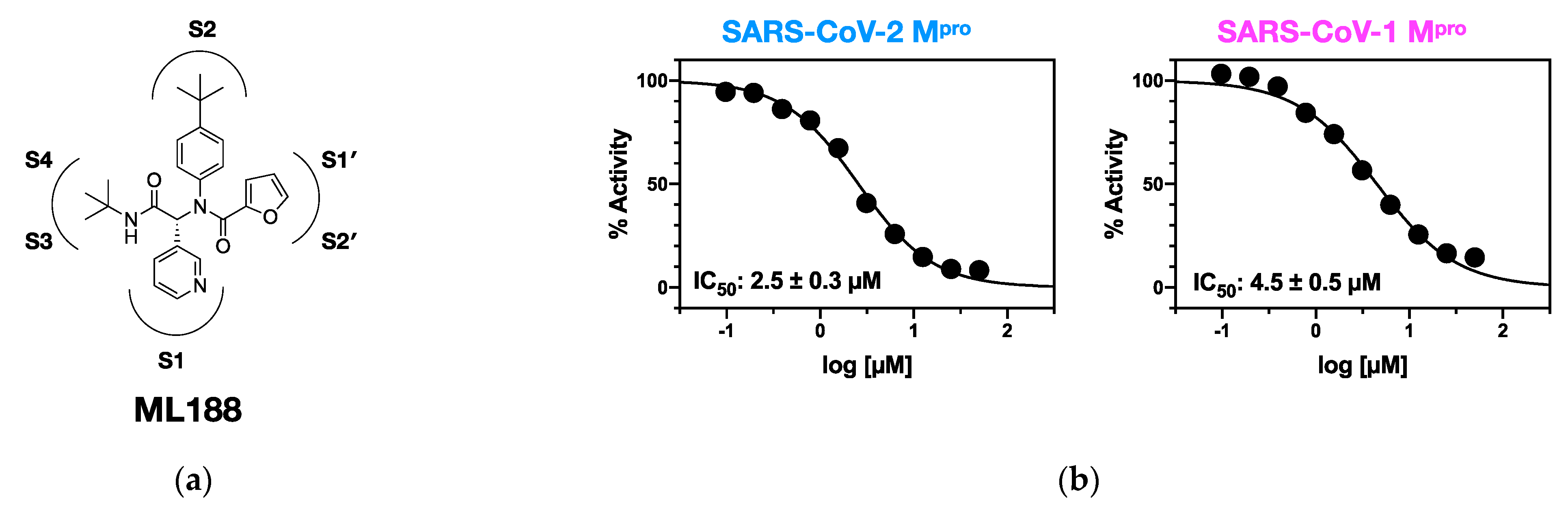
{kind=link}
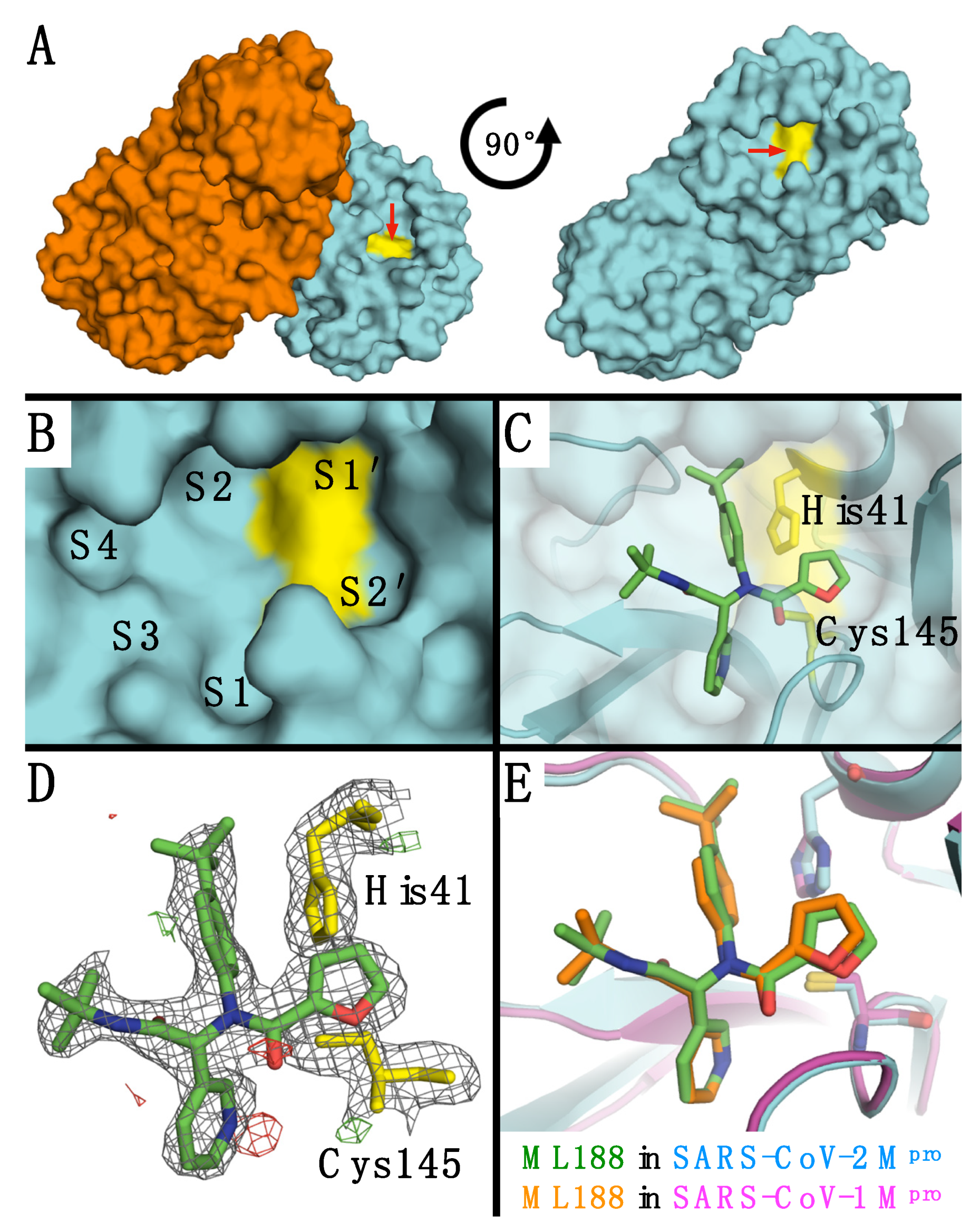
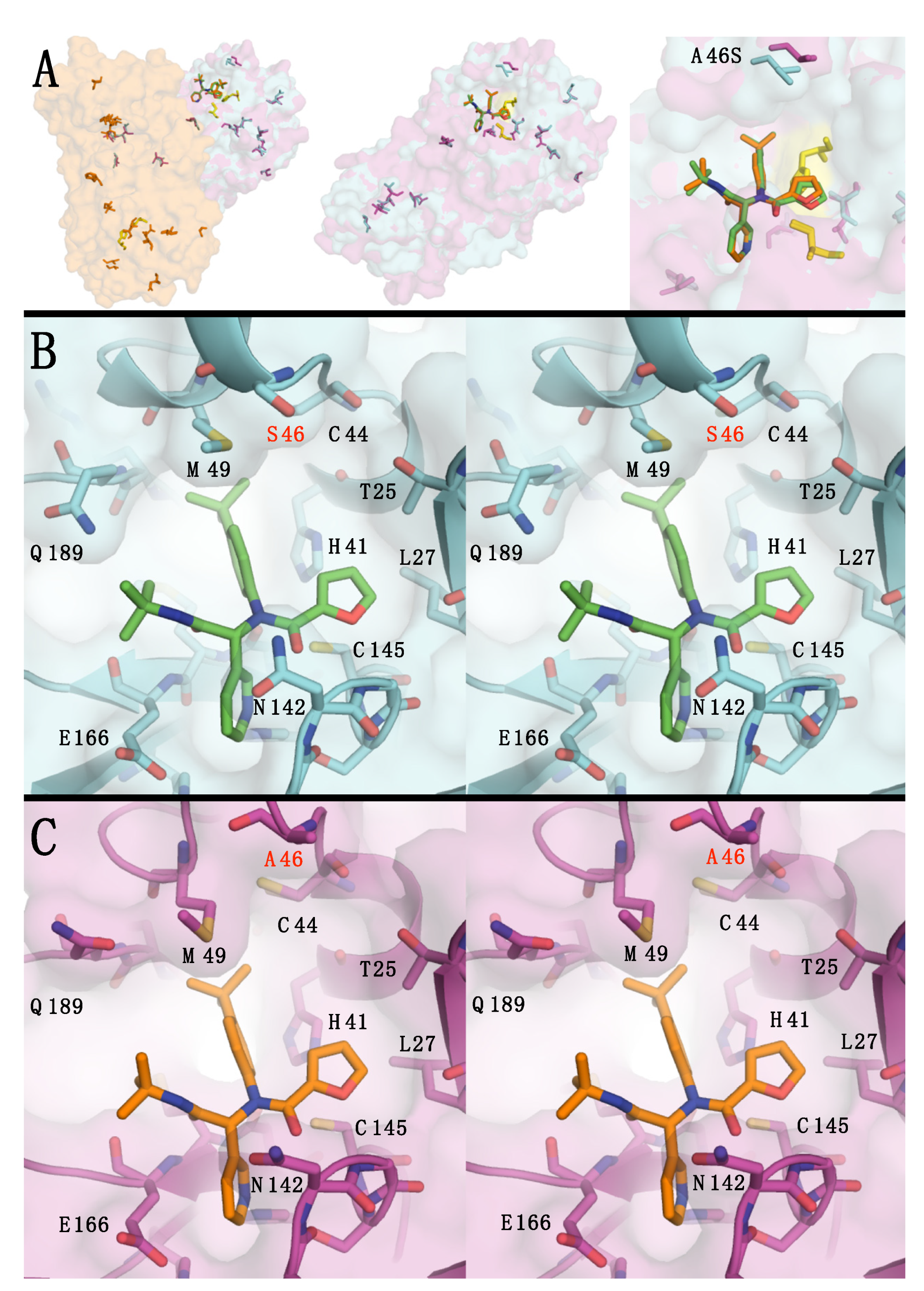{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification of SARS2-Mpro
2.2. MPro Inhibition Assay
2.3. Protein Crystallization
2.4. Data Collection and Structure Determination
2.5. Intermolecular vdW Contact Analysis of Crystal Structures
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. China Medical Treatment Expert Group for C. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Drosten, C.; Gunther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.E. The epidemiology of rheumatoid arthritis. Rheum. Dis. Clin. N. Am. 2001, 27, 269–281. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends. Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Bzowka, M.; Mitusinska, K.; Raczynska, A.; Samol, A.; Tuszynski, J.A.; Gora, A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, S.; Deng, W.; Zhang, Y.; Li, G.; Sun, J.; Zhao, W.; Guo, Y.; Yin, Z.; Li, D.; et al. Comprehensive Insights into the Catalytic Mechanism of Middle East Respiratory Syndrome 3C-Like Protease and Severe Acute Respiratory Syndrome 3C-Like Protease. ACS Catal. 2020, 10, 5871–5890. [Google Scholar] [CrossRef]
- Ragland, D.A.; Nalivaika, E.A.; Nalam, M.N.; Prachanronarong, K.L.; Cao, H.; Bandaranayake, R.M.; Cai, Y.; Kurt-Yilmaz, N.; Schiffer, C.A. Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J. Am. Chem. Soc. 2014, 136, 11956–11963. [Google Scholar] [CrossRef]
- Ragland, D.A.; Whitfield, T.W.; Lee, S.K.; Swanstrom, R.; Zeldovich, K.B.; Kurt-Yilmaz, N.; Schiffer, C.A. Elucidating the Interdependence of Drug Resistance from Combinations of Mutations. J. Chem. Theory Comput. 2017, 13, 5671–5682. [Google Scholar] [CrossRef] [PubMed]
- Henes, M.; Lockbaum, G.J.; Kosovrasti, K.; Leidner, F.; Nachum, G.S.; Nalivaika, E.A.; Lee, S.K.; Spielvogel, E.; Zhou, S.; Swanstrom, R.; et al. Picomolar to Micromolar: Elucidating the Role of Distal Mutations in HIV-1 Protease in Conferring Drug Resistance. ACS Chem. Biol. 2019, 14, 2441–2452. [Google Scholar] [CrossRef]
- Leidner, F.; Kurt Yilmaz, N.; Schiffer, C.A. Target-Specific Prediction of Ligand Affinity with Structure-Based Interaction Fingerprints. J. Chem. Inf. Model. 2019, 59, 3679–3691. [Google Scholar] [CrossRef]
- Whitfield, T.W.; Ragland, D.A.; Zeldovich, K.B.; Schiffer, C.A. Characterizing Protein-Ligand Binding Using Atomistic Simulation and Machine Learning: Application to Drug Resistance in HIV-1 Protease. J. Chem. Theory Comput. 2020, 16, 1284–1289. [Google Scholar] [CrossRef]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Abranyi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease as a Clinical Candidate for the Potential Treatment of COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M.; Tomar, S.; et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013, 56, 534–546. [Google Scholar] [CrossRef]
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, D.; Resnick, E.; Strain-Damerell, C.; et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. bioRxiv 2020. [Google Scholar] [CrossRef]
- St John, S.E.; Anson, B.J.; Mesecar, A.D. X-Ray Structure and Inhibition of 3C-like Protease from Porcine Epidemic Diarrhea Virus. Sci. Rep. 2016, 6, 25961. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 12, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 213–221. [Google Scholar] [CrossRef]
- Moriarty, N.W.; Grosse-Kunstleve, R.W.; Adams, P.D. electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 2009, 65 Pt 10, 1074–1080. [Google Scholar] [CrossRef]
- Brunger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein. Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Schrödinger. Schrödinger Release 2019-2; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Nalam, M.N.; Ali, A.; Altman, M.D.; Reddy, G.S.; Chellappan, S.; Kairys, V.; Ozen, A.; Cao, H.; Gilson, M.K.; Tidor, B.; et al. Evaluating the Substrate-Envelope Hypothesis: Structural Analysis of Novel HIV-1 Protease Inhibitors Designed to be Robust against Drug Resistance. J. Virol. 2010, 84, 368–378. [Google Scholar] [CrossRef]
- Tozser, J.; Oroszlan, S. Proteolytic events of HIV-1 replication as targets for therapeutic intervention. Curr. Pharm. Des. 2003, 9, 1803–1815. [Google Scholar] [CrossRef]
- Tozser, J.; Zahuczky, G.; Bagossi, P.; Louis, J.M.; Copeland, T.D.; Oroszlan, S.; Harrison, R.W.; Weber, I.T. Comparison of the substrate specificity of the human T-cell leukemia virus and human immunodeficiency virus proteinases. Eur. J. Biochem. 2000, 267, 6287–6295. [Google Scholar] [CrossRef]
- Tozser, J.; Bagossi, P.; Weber, I.T.; Louis, J.M.; Copeland, T.D.; Oroszlan, S. Studies on the symmetry and sequence context dependence of the HIV-1 proteinase specificity. J. Biol. Chem. 1997, 272, 16807–16814. [Google Scholar] [CrossRef] [PubMed]
- Tozser, J.; Weber, I.T.; Gustchina, A.; Blaha, I.; Copeland, T.D.; Louis, J.M.; Oroszlan, S. Kinetic and modeling studies of S3-S3′ subsites of HIV proteinases. Biochemistry 1992, 31, 4793–4800. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.M.; Copeland, T.D.; Oroszlan, S. In situ processing of a retroviral nucleocapsid protein by the viral proteinase. Protein Eng. 1991, 4, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Tozser, J.; Gustchina, A.; Weber, I.T.; Blaha, I.; Wondrak, E.M.; Oroszlan, S. Studies on the role of the S4 substrate binding site of HIV proteinases. FEBS Lett. 1991, 279, 356–360. [Google Scholar] [CrossRef]
- Bagossi, P.; Cheng, Y.S.; Oroszlan, S.; Tozser, J. Comparison of the specificity of homo- and heterodimeric linked HIV-1 and HIV-2 proteinase dimers. Protein Eng. 1998, 11, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Tozser, J.; Blaha, I.; Copeland, T.D.; Wondrak, E.M.; Oroszlan, S. Comparison of the HIV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol polyproteins. FEBS Lett. 1991, 281, 77–80. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Kurt Yilmaz, N.; Thompson, P.R.; Schiffer, C.A. Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses 2021, 13, 174. https://doi.org/10.3390/v13020174
Lockbaum GJ, Reyes AC, Lee JM, Tilvawala R, Nalivaika EA, Ali A, Kurt Yilmaz N, Thompson PR, Schiffer CA. Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses. 2021; 13(2):174. https://doi.org/10.3390/v13020174
Chicago/Turabian StyleLockbaum, Gordon J., Archie C. Reyes, Jeong Min Lee, Ronak Tilvawala, Ellen A. Nalivaika, Akbar Ali, Nese Kurt Yilmaz, Paul R. Thompson, and Celia A. Schiffer. 2021. "Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188" Viruses 13, no. 2: 174. https://doi.org/10.3390/v13020174
APA StyleLockbaum, G. J., Reyes, A. C., Lee, J. M., Tilvawala, R., Nalivaika, E. A., Ali, A., Kurt Yilmaz, N., Thompson, P. R., & Schiffer, C. A. (2021). Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses, 13(2), 174. https://doi.org/10.3390/v13020174