
Nuclear Import of Adeno-Associated Viruses Imaged by High-Speed Single-Molecule Microscopy


Abstract
1. Introduction
1.1. Overview of the NPC and Nuclear Transport of Viruses
1.2. Live Cell Imaging with High Spatial Resolution Is Needed to Detail Viral Nuclear Transport Kinetics through the NPC
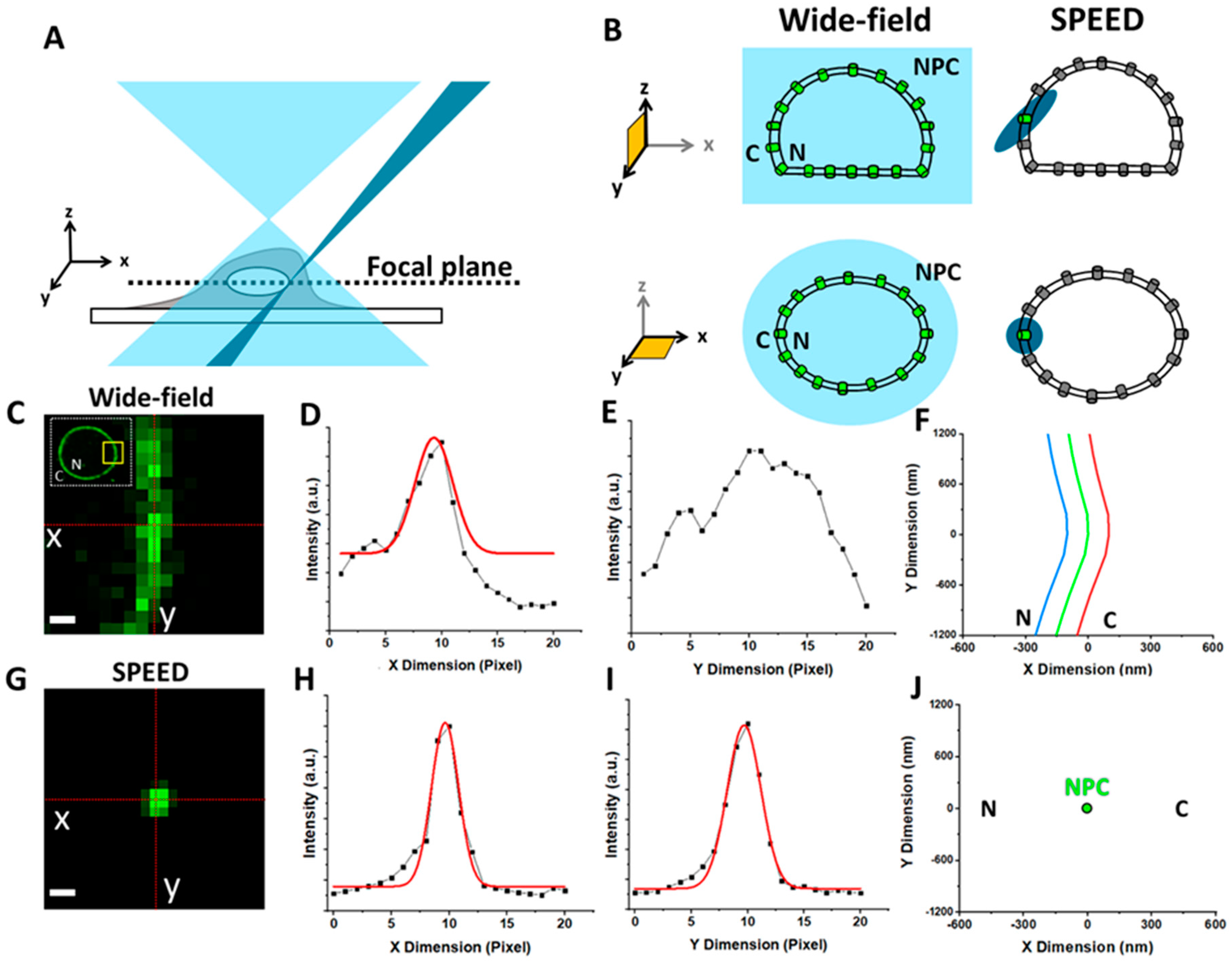
1.3. Overview of Fluorescence Microscopy Techniques Applied to Study Viral–NPC Interactions
2. Description of Methods
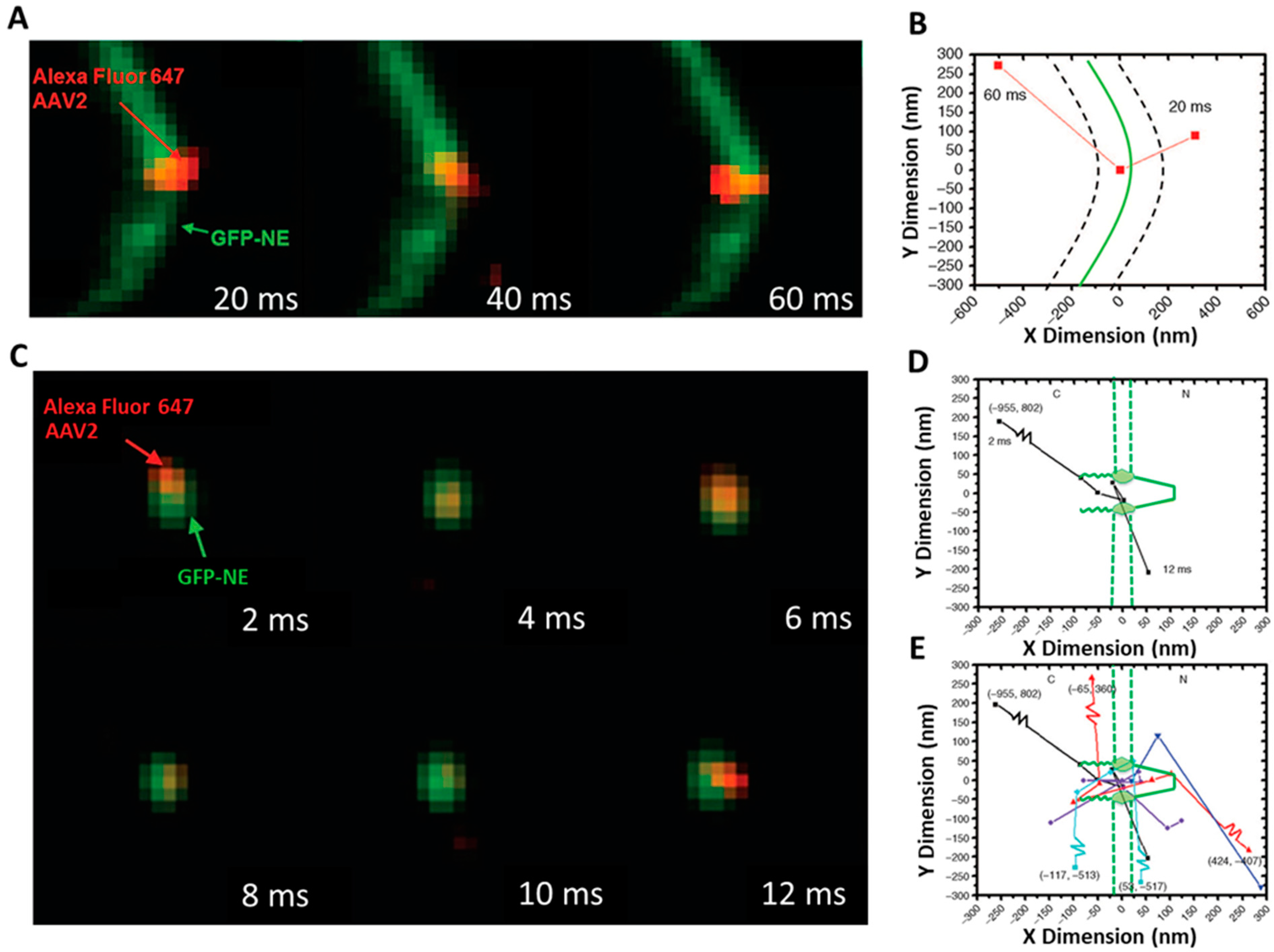
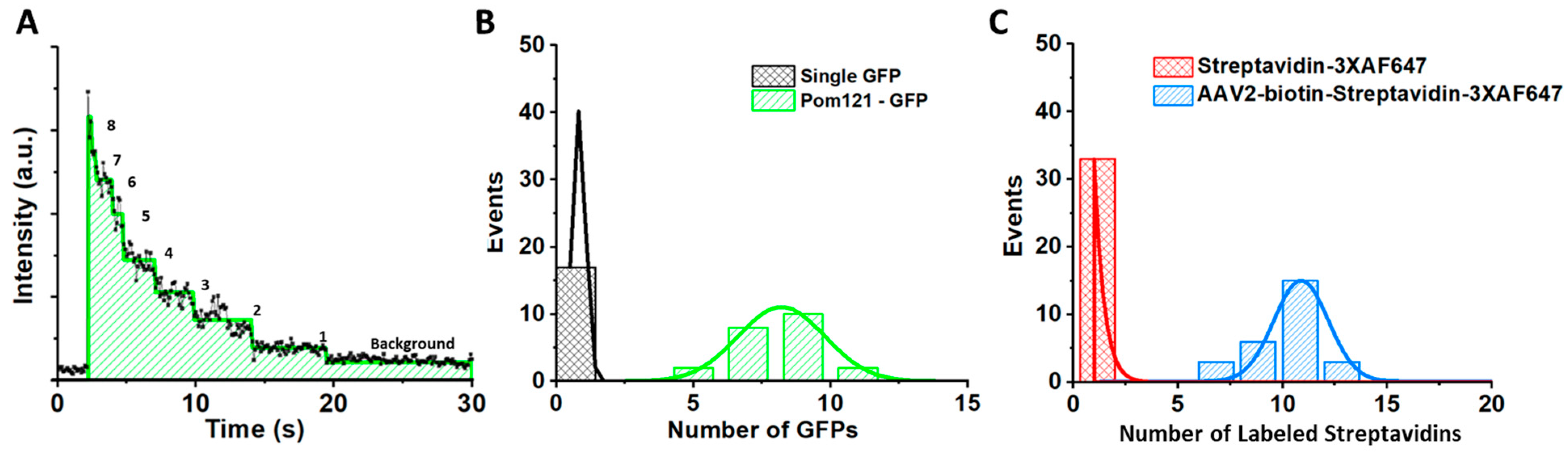
SPEED Microscopy Illuminates Individual AAV Molecule Transport through Single NPCs
3. Discussion
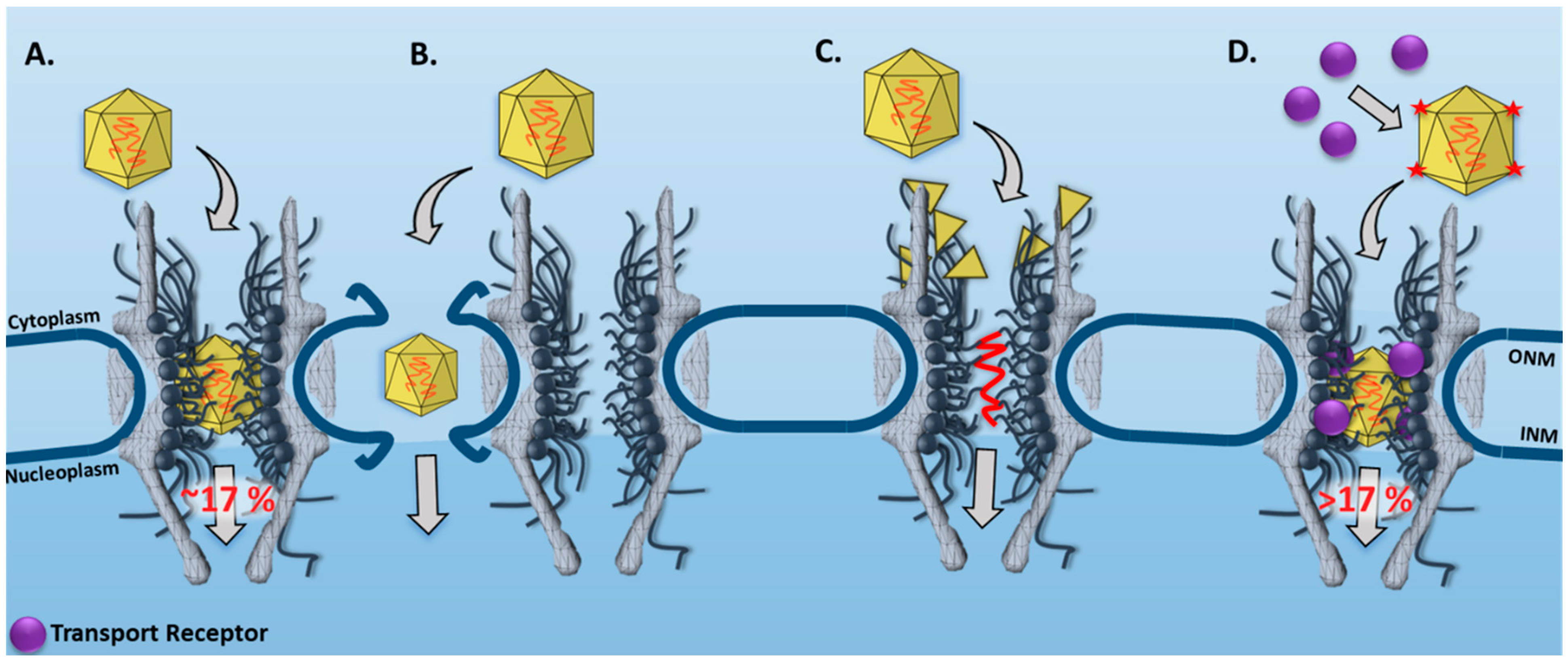
3.1. New Features for the Nuclear Transport of AAV Particles Obtained by SPEED Microscopy
3.2. Limitations of SPEED Microscopy
4. Description of the Equipment
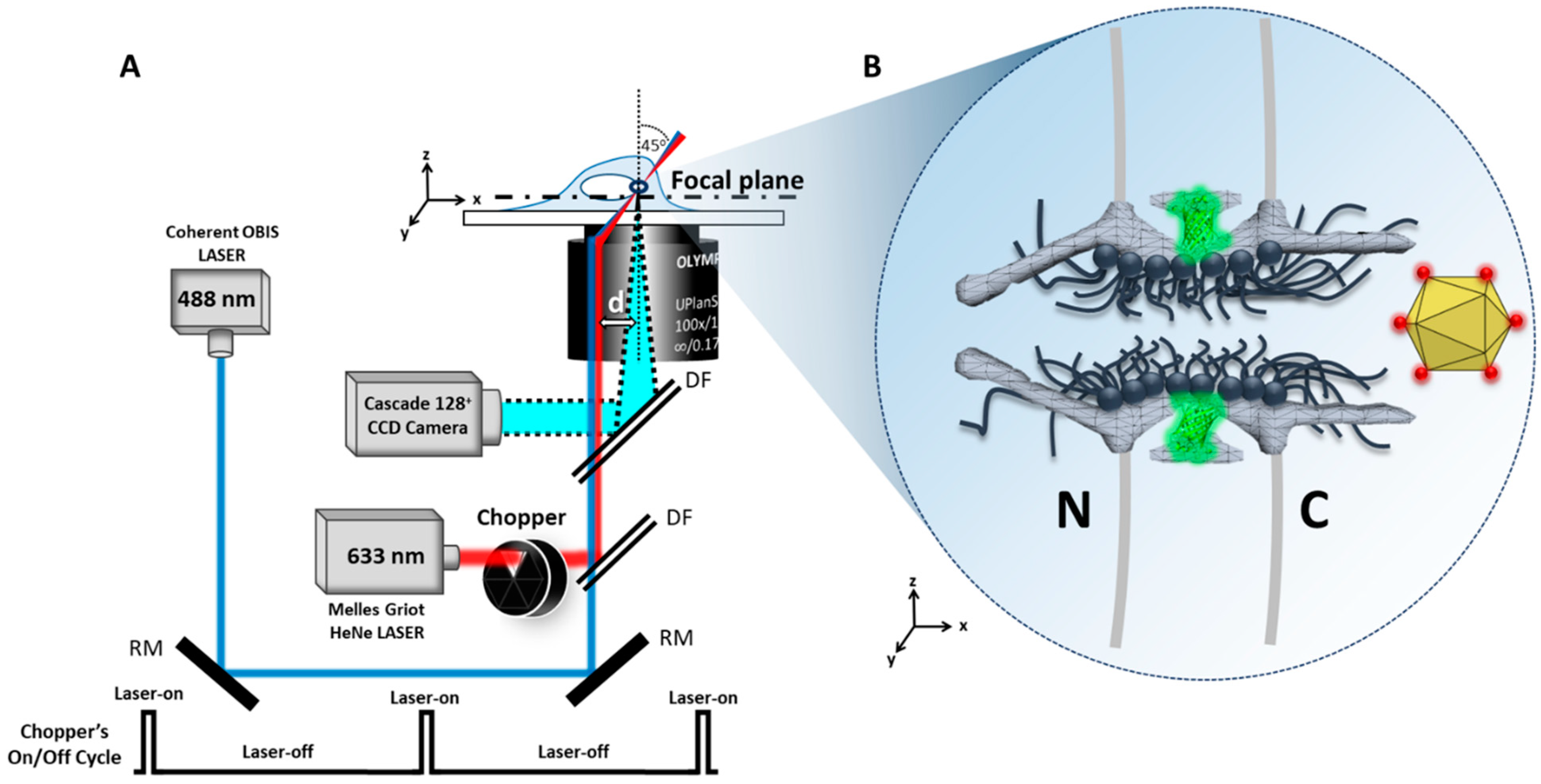
4.1. Microscopy
4.2. Laser Illumination and Filter Setting
4.3. CCD Camera
4.4. Imaging Software
5. Experimental Procedures
5.1. Preparation of AAV Particles
5.1.1. Production of AAV Particles
- Prepare purified plasmids pAAV-GOI, pRC, pHelper, and pBirA-EGFP. The pRC has the AVI peptide sequence inserted into AA139–140 of the basic region (BR1) location.
- Culture HEK293A cells in DMEM high glucose with L-glutamine and 1 mM pyruvate (Gibco), supplemented by 10% (v/v) Fetal Calf Serum (FBS) and 1% (v/v) Antibiotic–Antimycotic. Plate and incubate HEK293A cells at 37 °C within a 5% CO2 incubator until 70–80% confluent.
- With calcium phosphate transfection, insert pAAV-GOI, pRC, pHelper, and pBirA-EGFP into the HEK293A cells. First, mix a 1:1:1:2 molar ratio of pAAV-GOI:pRC:pHelper:pBirA-EGFP for a total DNA concentration of 25 µg. Adjust the total volume of DNA to 25 µL.
- Mix 30 µL of CaCl2 (2.5 M), 245 µL H2O, and 25 µL of the DNA. Add the DNA mixture dropwise to bubbling 300 µL of 2X HEPES-buffered saline (HBS; 50 mM HEPES [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid], 280 mM NaCl, and 1.5 mM Na2HPO4, pH 7.05) [74]. To bubble the HEPES-buffered saline, use a Pasteur pipette to slowly inject air into the solution as the DNA mixture is added.
- Vortex the mixture and allow it to sit for 20 min. Confirm that the precipitate is fine with little aggregation. If aggregation does occur, adjust the pH of 2X HEPES to produce a fine precipitate.
- Add this mixture dropwise to the HEK293A cells and incubate the cells at 37 °C within a 5% CO2 incubator for at least 16 h undisturbed.
- Replace the cell culture media and incubate for an additional 24–48 h.
- rAAV is nonlytic, so the media will have low titer of virus. After incubation, gently knock the plate to lift the cells. Alternatively, add EDTA (0.5 M) to help lift the cells. If a higher titer is desired, retain cell media and add to the AAV precipitation step.
- To ensure effective labeling of biotin on AAV2 capsid proteins, fluorescence-activated cell sorting (FACS) is used to separate the transfected HEK293A cells expressing EGFP from the other cells.
- Centrifuge the fluorescently labeled cells at 500× g for 5 min. Then, resuspend in 1 mL of phosphate-buffered saline (PBS; pH 7.4).
- Lyse the cells using a freeze/thaw method of 10 min in liquid nitrogen or a dry ice–ethanol bath, then transfer to a 37 °C water bath until the cellular mixture is completely thawed. Repeat the freeze/thaw three more times (HINT 1).
- After the last thaw at 37 °C, add 50 U/mL benzonase and 10 U/mL RNase I to the virus-released solution. Incubate for 30 min at 37 °C in a water bath. Then, add 0.5% sodium deoxycholate and incubate for an additional 30 min.
- Centrifuge the mixture at 10,000× g for 10 min and save the supernatant. Add PEG 8000 and NaCl for a final concentration of 8% PEG 8000 and 0.5 M NaCl. Then, incubate on ice for 60 min.
- Centrifuge AAV particles at 2000× g for 30 min at 4 °C. Then, resuspend the pellet in a low volume of HBS (pH 8.0). The crude pellet can be stored at −80 °C.
5.1.2. Purification of AAV Particles
- Add an equal volume of chloroform to the crude rAAV pellet. Vortex vigorously for 2 min until a homogenous mixture is obtained. Then, centrifuge for 5 min at 370× g and retain the aqueous phase. In a sterile environment, vent the aqueous phase for 30 min. This will evaporate the remaining chloroform.
- Dilute aqueous phase into 10%PEG8000/13.2% (NH4)2SO4 ((w/w), pH 8.0) (HINT 2).
- Let the mixture incubate for 15–30 min at room temperature, then centrifuge at 3000× g for 15 min. Then, carefully draw out the clear bottom phase with a needle. The bottom clear phase is the virus-containing phase.
- Concentrate this phase with centrifugal filters (Amicon Ultra-0.5 mL Centrifugal Filters; Ultra 50K device). At room temperature, centrifuge the sample for 20 min at 14,000× g. Rinse the sample twice with a low-salt solution (NaCl (10 mM), pH 7.4).
- Store the final concentrated rAAV stock at −20 °C in PBS (pH 7.4) or minimum essential media (MEM; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) with 0.001% (v/v) pluronic F68 (Gibco).
5.1.3. Labeling of AAV Particles
- At a concentration ratio of 40:1 (streptavidin–biotin), incubate the Alexa Fluor 647-labeled streptavidin with biotin-labeled AAV2 particles at 4 °C while agitating for 1 h while covered from light.
- Isolate the labeled AAV2 particles from free streptavidin by centrifugal filtration (Amicon Ultra-0.5 mL Centrifugal Filters). We recommend the Amicon Ultra 50K device.
- At room temperature, centrifuge the sample for 20 min at 14,000× g. Rinse the sample twice with PBS (pH 7.4). Then, measure the labeling efficiency of the AAV2 particles, as shown in Section 5.5.
5.2. Preparation of Purified Proteins
5.2.1. Bacterial Transformation and Expression
- Transform BL21 (DE3) (New England BioLabs Inc., Ipswich, MA, USA) E. coli cells with ~0.1–100 ng of Impβ1 plasmid DNA through a heat shock at 42 °C for 10 s.
- Incubate the transformed BL21 (DE3) cells for 60 min at 37 °C in a 19:1 dilution of Super Optimal Broth (SOC, 0.5% Yeast Extract, 2% Tryptone, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, 20mM Glucose) Media to transform BL21 (DE3) cells.
- Spread 50 µL of the mixture onto ampicillin (100 mg/mL) agar plates and incubate at 37 °C overnight (12–14 h) (HINT 3).
- Create a starter culture from a single bacterial colony in 5 mL of Lysogeny Broth (LB) with the addition of 5 μL of ampicillin (100 mg/mL) and grow aerobically with shaking overnight (12–14 h) at 37 °C at 225 rpm.
- Transfer the 5 mL saturated starter culture to 1 L of LB media with 1 mL ampicillin (100 mg/mL). Shake the mixture at 37 °C until an OD 600 nm of ~0.6 is reached, usually 5–6 h. Add 1 mL of IPTG (1 M) to activate protein production and incubate overnight at 30 °C (HINT 4).
5.2.2. Protein Purification
5.2.3. Protein Characterization
5.2.4. Protein Desalting and Storage
- To further isolate Impβ1 by centrifugal filters (Amicon Ultra-0.5 mL Centrifugal Filters), select a filter that will collect the target protein in the loading chamber while the contaminants elute through. For Impβ1, we used an Amicon Ultra 50K device, since Impβ1 has a molecular weight of ~97 kDa.
- At room temperature, centrifuge the sample for 20 min at 14,000× g. Rinse the sample twice with a low-salt solution (NaCl (10 mM), pH 7.4).
- Recover the concentrated target protein by inverting the filter into a clean tube and centrifuge for 2 min at 1000× g.
- Resuspend the protein in one of the following solutions for storage [79]:
- for 24 h at 4 °C, PBS (pH 7.4);
- for one week at 4 °C, PBS (pH 7.4) with a bacteriostatic agent (0.1% (w/v) sodium azide)
- from one week to several months at −20 °C, PBS (50% (w/v) glycerol, 10 mg/mL BSA, 1 mM DDT, pH 7.4) (HINT 6);
- for months to years at −80 °C, PBS (50% (w/v) glycerol).
5.3. Preparation of a Cell System
5.3.1. Preparation of the Live Cell System
5.3.2. Preparation of the Permeabilized Cell System
5.4. SPEED Microscopy Tracking of Single Alexa Fluor 647-Labeled AAV through a Single GFP-Labeled NPC in Live and Digitonin-Permeabilized Cells
5.4.1. Tracking of Single AAV Particles in Live Cells
5.4.2. Tracking of Single AAV Particles in Permeabilized Cells
5.5. Determining the Copy Number of POM121-GFP and the Labeling Efficiency of the Fluorescently Labeled AAV Particles
5.6. Localization of the NE and a Single NPC on the NE
5.7. Localization Precision of Isolated Fluorescent Spots
5.8. Calculation of Transport Time and Transport Efficiency
6. Hints
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cohen, S.; Au, S.; Panté, N. How viruses access the nucleus. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Rout, M.P.; Blobel, G. Isolation of the yeast nuclear pore complex. J. Cell Biol. 1993, 123, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Görlich, D.; Kutay, U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999, 15, 607–660. [Google Scholar] [CrossRef]
- Macara, I.G. Transport into and out of the nucleus. Microbiol. Mol. Biol. Rev. 2001, 65, 570–594. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Distinct, but not completely separate spatial transport routes in the nuclear pore complex. Nucleus 2013, 4, 166–175. [Google Scholar] [CrossRef]
- Maimon, T.; Elad, N.; Dahan, I.; Medalia, O. The human nuclear pore complex as revealed by cryo-electron tomography. Structure 2012, 20, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Kelich, J.M.; Junod, S.L.; Yang, W. Super-resolution mapping of scaffold nucleoporins in the nuclear pore complex. J. Cell Sci. 2017, 130, 1299–1306. [Google Scholar] [CrossRef]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef]
- Alber, F.; Dokudovskaya, S.; Veenhoff, L.M.; Zhang, W.; Kipper, J.; Devos, D.; Suprapto, A.; Karni-Schmidt, O.; Williams, R.; Chait, B.T.; et al. The molecular architecture of the nuclear pore complex. Nat. Cell Biol. 2007, 450, 695–701. [Google Scholar] [CrossRef]
- Vasu, S.K.; Forbes, D.J. Nuclear pores and nuclear assembly. Curr. Opin. Cell Biol. 2001, 13, 363–375. [Google Scholar] [CrossRef]
- Mattaj, I.W.; Englmeier, L. Nucleocytoplasmic transport: The soluble phase. Annu. Rev. Biochem. 1998, 67, 265–306. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Goryaynov, A.; Yang, W. Super-resolution 3D tomography of interactions and competition in the nuclear pore complex. Nat. Struct. Mol. Biol. 2016, 23, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Goryaynov, A.; Sarma, A.; Yang, W. Self-regulated viscous channel in the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2012, 109, 7326–7331. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.Y.H.; Huang, N.-P.; Köser, J.; Deng, J.; Lau, K.H.A.; Schwarz-Herion, K.; Fahrenkrog, B.; Aebi, U. Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 2006, 103, 9512–9517. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Dougherty, M.; Jakana, J.; He, J.; Rixon, F.J.; Chiu, W. Seeing the herpesvirus capsid at 8.5 A. Science 2000, 288, 877–880. [Google Scholar] [CrossRef]
- Ojala, P.M.; Sodeik, B.; Ebersold, M.W.; Kutay, U.; Helenius, A. Herpes simplex virus type 1 entry into host cells: Reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 2000, 20, 4922–4931. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef]
- Copeland, A.M.; Newcomb, W.W.; Brown, J.C. Herpes simplex virus replication: Roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 2008, 83, 1660–1668. [Google Scholar] [CrossRef]
- Pasdeloup, D.; Blondel, D.; Isidro, A.; Rixon, F.J. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL. J. Virol. 2009, 83, 6610–6623. [Google Scholar] [CrossRef]
- Reddy, V.S.; Natchiar, S.; Gritton, L.; Mullen, T.-M.; Stewart, P.; Nemerow, G.R. Crystallization and preliminary X-ray diffraction analysis of human adenovirus. Virology 2010, 402, 209–214. [Google Scholar] [CrossRef]
- Hindley, C.E.; Lawrence, F.J.; Matthews, D.A. A role for transportin in the nuclear import of adenovirus core proteins and DNA. Traffic 2007, 8, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R.H.; Kawaoka, Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006, 439, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Glass, B.; Hagen, W.J.H.; Kräusslich, H.-G.; Briggs, J.A.G. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 2016, 354, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-N.; Millman, I.; O’Connell, A.; Aronoff, A.; Gault, H.; Blumberg, B.S. Virus-like particles in Australia antigen-associated hepatitis. An immunoelectron microscopic study of human liver. Am. J. Pathol. 1972, 67, 453–470. [Google Scholar]
- Rayaprolu, V.; Kruse, S.; Kant, R.; Venkatakrishnan, B.; Movahed, N.; Brooke, D.; Lins, B.; Bennett, A.; Potter, T.; McKenna, R.; et al. Comparative analysis of adeno-associated virus capsid stability and dynamics. J. Virol. 2013, 87, 13150–13160. [Google Scholar] [CrossRef]
- Jenkins, Y.; McEntee, M.; Weis, K.; Greene, W.C. Characterization of HIV-1 Vpr nuclear import: Analysis of signals and pathways. J. Cell Biol. 1998, 143, 875–885. [Google Scholar] [CrossRef]
- Haffar, O.K.; Popov, S.; Dubrovsky, L.; Agostini, I.; Tang, H.; Pushkarsky, T.; Nadler, S.G.; Bukrinsky, M. Two nuclear localization signals in the HIV-1 matrix protein regulate nuclear import of the HIV-1 pre-integration complex. J. Mol. Biol. 2000, 299, 359–368. [Google Scholar] [CrossRef]
- Gallay, P.; Hope, T.; Chin, D.; Trono, D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 9825–9830. [Google Scholar] [CrossRef]
- Popov, S.; Rexach, M.; Zybarth, G.; Reiling, N.; Lee, M.; Ratner, L.; Lane, C.M.; Moore, M.S.; Blobel, G.; Bukrinsky, M. Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 1998, 17, 909–917. [Google Scholar] [CrossRef]
- Ao, Z.; Huang, G.; Yao, H.; Xu, Z.; LaBine, M.; Cochrane, A.W.; Yao, X. Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication. J. Biol. Chem. 2007, 282, 13456–13467. [Google Scholar] [CrossRef]
- Wang, P.; Palese, P.; O’Neill, R.E. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 1997, 71, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Jaskunas, R.; Blobel, G.; Palese, P.; Moroianu, J. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J. Biol. Chem. 1995, 270, 22701–22704. [Google Scholar] [CrossRef] [PubMed]
- Kelich, J.M.; Ma, J.; Dong, B.; Wang, Q.; Chin, M.; Magura, C.M.; Xiao, W.; Yang, W. Super-resolution imaging of nuclear import of adeno-associated virus in live cells. Mol. Ther. Methods Clin. Dev. 2015, 2, 15047. [Google Scholar] [CrossRef]
- Popa-Wagner, R.; Sonntag, F.; Schmidt, K.; King, J.; Kleinschmidt, J.A. Nuclear translocation of adeno-associated virus type 2 capsid proteins for virion assembly. J. Gen. Virol. 2012, 93, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Akiba, T.; Nakayama, H.; Miyazaki, Y.; Kanno, A.; Ishii, M.; Ohori, H. Relationship between the replication of hepatitis B virus and the localization of virus nucleocapsid antigen (HBcAg) in hepatocytes. J. Gen. Virol. 1987, 68, 871–877. [Google Scholar] [CrossRef]
- Kann, M.; Sodeik, B.; Vlachou, A.; Gerlich, W.H.; Helenius, A. phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J. Cell Biol. 1999, 145, 45–55. [Google Scholar] [CrossRef]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Panté, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Panté, N. Pushing the envelope: Microinjection of Minute virus of mice into Xenopus oocytes causes damage to the nuclear envelope. J. Gen. Virol. 2005, 86, 3243–3252. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Van Oijen, A.M.; Köhler, J.; Schmidt, J.; Muller, M.; Brakenhoff, G.J. Far-field fluorescence microscopy beyond the diffraction limit. J. Opt. Soc. Am. A 1999, 16, 909–915. [Google Scholar] [CrossRef]
- Klar, T.A.; Hell, S.W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999, 24, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Engel, E.; Hell, S.W. Breaking Abbe’s diffraction resolution limit in fluorescence microscopy with stimulated emission depletion beams of various shapes. Phys. Rev. E 2001, 64, 066613. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.I.; Churchman, L.S.; Spudich, J.A.; Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 2010, 7, 377–381. [Google Scholar] [CrossRef]
- Erni, R.; Rossell, M.D.; Kisielowski, C.; Dahmen, U. Atomic-resolution imaging with a sub-50-pm electron probe. Phys. Rev. Lett. 2009, 102, 096101. [Google Scholar] [CrossRef]
- Milazzo, A.-C.; Cheng, A.; Moeller, A.; Lyumkis, D.; Jacovetty, E.; Polukas, J.; Ellisman, M.H.; Xuong, N.-H.; Carragher, B.; Potter, C.S. Initial evaluation of a direct detection device detector for single particle cryo-electron microscopy. J. Struct. Biol. 2011, 176, 404–408. [Google Scholar] [CrossRef]
- Bammes, B.E.; Rochat, R.H.; Jakana, J.; Chen, D.H.; Chiu, W. Direct electron detection yields cryo-EM reconstructions at resolutions beyond 3/4 Nyquist frequency. J. Struct. Biol. 2012, 177, 589–601. [Google Scholar] [CrossRef]
- Featherstone, C.; Darby, M.K.; Gerace, L. A monoclonal antibody against the nuclear pore complex inhibits nucleocytoplasmic transport of protein and RNA in vivo. J. Cell Biol. 1988, 107, 1289–1297. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.L.; Longshaw, S.V.; Kirby, I.; Santis, G.; Tobin, M.J.; Clarke, D.T.; Jones, G.R. Adenovirus type-5 entry and disassembly followed in living cells by FRET, fluorescence anisotropy, and FLIM. Biophys. J. 2004, 87, 1316–1327. [Google Scholar] [CrossRef]
- Babcock, H.; Chen, C.; Zhuang, X. Using single-particle tracking to study nuclear trafficking of viral genes. Biophys. J. 2004, 87, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 imaging reveals progression of infection through CA-dependent steps of docking at the nuclear pore, uncoating, and nuclear transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef] [PubMed]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.R.; Newmeyer, D.D.; Price, T.M.; Forbes, D.J. Inhibition of in vitro nuclear transport by a lectin that binds to nuclear pores. J. Cell Biol. 1987, 104, 189–200. [Google Scholar] [CrossRef]
- Li, B.; Kohler, J.J. Glycosylation of the nuclear pore. Traffic 2014, 15, 347–361. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Ma, J.; Yang, W. Three-dimensional distribution of transient interactions in the nuclear pore complex obtained from single-molecule snapshots. Proc. Natl. Acad. Sci. USA 2010, 107, 7305–7310. [Google Scholar] [CrossRef]
- Daigle, N.; Beaudouin, J.; Hartnell, L.; Imreh, G.; Hallberg, E.; Lippincott-Schwartz, J.; Ellenberg, J. Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J. Cell Biol. 2001, 154, 71–84. [Google Scholar] [CrossRef]
- Kubitscheck, U.; Wedekind, P.; Zeidler, O.; Grote, M.; Peters, R. Single nuclear pores visualized by confocal microscopy and image processing. Biophys. J. 1996, 70, 2067–2077. [Google Scholar] [CrossRef]
- Li, Y.; Junod, S.L.; Ruba, A.; Kelich, J.M.; Yang, W. Nuclear export of mRNA molecules studied by SPEED microscopy. Methods 2019, 153, 46–62. [Google Scholar] [CrossRef]
- Ma, J.; Liu, Z.; Michelotti, N.; Pitchiaya, S.; Veerapaneni, R.; Androsavich, J.R.; Walter, N.G.; Yang, W. High-resolution three-dimensional mapping of mRNA export through the nuclear pore. Nat. Commun. 2013, 4, 2414. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, G. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294, 1929–1932. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.-J.; Samulski, R.J. Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. J. Virol. 2012, 86, 10462–10473. [Google Scholar] [CrossRef] [PubMed]
- Ribbeck, K.; Görlich, D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J. 2002, 21, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.-C.; Fu, G.; Zilman, A.; Musser, S.M. Large cargo transport by nuclear pores: Implications for the spatial organization of FG-nucleoporins. EMBO J. 2013, 32, 3220–3230. [Google Scholar] [CrossRef]
- Junod, S.L.; Kelich, J.M.; Ma, J.; Yang, W. Nucleocytoplasmic transport of intrinsically disordered proteins studied by high-speed super-resolution microscopy. Protein Sci. 2020, 29, 1459–1472. [Google Scholar] [CrossRef]
- Li, Y.; Tingey, M.; Ruba, A.; Yang, W. High-speed super-resolution imaging of rotationally symmetric structures using SPEED microscopy and 2D-to-3D transformation. Nat. Protoc. 2021, 16, 532–560. [Google Scholar] [CrossRef]
- Gelles, J. Gelles-Brandeis. 2014. Available online: https://github.com/gelles-brandeis/Glimpse (accessed on 12 May 2020).
- Herbert, A. Single Molecule Light Microscopy ImageJ Plugins. 2014. Available online: http://www.sussex.ac.uk/gdsc/intranet/microscopy/UserSupport/AnalysisProtocol/imagej/smlm_plugins/ (accessed on 12 May 2020).
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-associated defective virus particles. Science 1965, 149, 754–755. [Google Scholar] [CrossRef]
- Hoggan, M.D.; Blacklow, N.R.; Rowe, W.P. Studies of small DNA viruses found in various adenovirus preparations: Physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. USA 1966, 55, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.J. Adeno-associated virus and the development of adeno-associated virus vectors: A historical perspective. Mol. Ther. 2004, 10, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Elliger, S.; Elliger, C.; Podsakoff, G.; Villarreal, L.; Kurtzman, G.J.; Iwaki, Y.; Colosi, P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998, 5, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Zhou, S.; Nakai, H.; Thomas, C.E.; Storm, T.A.; Fuess, S.; Matsushita, T.; Allen, J.; Surosky, R.; Lochrie, M.; et al. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood 2003, 102, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.A.; Hoggan, M.D.; Shatkin, A.J. Nucleic acid from an adeno-associated virus: Chemical and physical studies. Proc. Natl. Acad. Sci. USA 1966, 56, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Strobel, B.; Miller, F.D.; Rist, W.; Lamla, T. Comparative analysis of cesium chloride- and iodixanol-based purification of recombinant adeno-associated viral vectors for preclinical applications. Hum. Gene Ther. Methods 2015, 26, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 2008, 8, 921–929. [Google Scholar] [CrossRef]
- Fairhead, M.; Howarth, M. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 2015, 1266, 171–184. [Google Scholar]
- Alberghina, D.; Casella, S.; Giannetto, C.; Marafioti, S.; Piccione, G. Effect of storage time and temperature on the total protein concentration and electrophoretic fractions in equine serum. Can. J. Veter. Res. 2013, 77, 293–296. [Google Scholar]
- Bader, M.F.; Thiersé, D.; Aunis, D.; Ahnert-Hilger, G.; Gratzl, M. Characterization of hormone and protein release from alpha-toxin-permeabilized chromaffin cells in primary culture. J. Biol. Chem. 1986, 261, 5777–5783. [Google Scholar] [CrossRef]
- GDSC Single Molecule Light Microscopy (SMLM) ImageJ Plugins. Available online: https://gdsc-smlm.readthedocs.io/en/latest/ (accessed on 12 May 2020).
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef]
- Quan, T.; Zeng, S.; Huang, Z.-L. Localization capability and limitation of electron-multiplying charge-coupled, scientific complementary metal-oxide semiconductor, and charge-coupled devices for superresolution imaging. J. Biomed. Opt. 2010, 15, 066005. [Google Scholar] [CrossRef]
- Von Diezmann, A.; Shechtman, Y.; Moerner, W.E. Three-dimensional localization of single molecules for super-resolution imaging and single-particle tracking. Chem. Rev. 2017, 117, 7244–7275. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; El-Gohary, Y.; Prasadan, K.; Shiota, C.; Xiao, X.; Wiersch, J.; Paredes, J.; Tulachan, S.; Gittes, G. Rapid and simplified purification of recombinant adeno-associated virus. J. Virol. Methods 2012, 183, 139–146. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Microscopy Technique | Main Observations/Conclusions | References |
|---|---|---|---|
| HSV | Confocal Laser Scanning Microscopy | In permeabilized fixed cells, the interaction between HSV-1 capsid protein pUL25 and Nup214 was identified as an important step for viral genome delivery into the nucleus. | [19] |
| Adenovirus | Fluorescence Resonance Energy Transfer (FRET) Microscopy | In live cells, the disassembly of adenovirus 5 viral capsids was quantified by a progressive decrease in FRET signal after the capsid docked to the NPC. Two major decreases occurred, suggesting a docked capsid half-life of 3 and 60 min. | [50] |
| Influenza | Epi-Fluorescence and Single-Particle Tracking | In live cells, the time trajectories of single vRNPs revealed dissociation rate constants ranging from 0.01 to 1 s−1 for binding between the vRNPs and the nuclear envelope (NE). In addition, a significant reduction in vRNP nuclear localization was observed in the presence of anti-NPC or wheat germ agglutinin (WGA), suggesting the NPC to be the primary transport route of vRNPs. | [51] |
| HIV | Super-Resolution Stimulated Emission Depletion (STED) Microscopy | In live cells, the HIV-1 nucleocapsid has been shown to dock at the NE, then import the PIC into the nucleus. | [52] |
| HBV | Confocal Laser Scanning Microscopy | In permeabilized cells, the transport of intact HBV capsids into the nucleus is facilitated by Impα and Impβ1. | [53] |
| AAV | SPEED Microscopy | In live cells, single intact AAV2 capsids were found to transport through single NPCs into the nucleus with a nuclear import success rate of ~17%. | [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Junod, S.L.; Saredy, J.; Yang, W. Nuclear Import of Adeno-Associated Viruses Imaged by High-Speed Single-Molecule Microscopy. Viruses 2021, 13, 167. https://doi.org/10.3390/v13020167
Junod SL, Saredy J, Yang W. Nuclear Import of Adeno-Associated Viruses Imaged by High-Speed Single-Molecule Microscopy. Viruses. 2021; 13(2):167. https://doi.org/10.3390/v13020167
Chicago/Turabian StyleJunod, Samuel L., Jason Saredy, and Weidong Yang. 2021. "Nuclear Import of Adeno-Associated Viruses Imaged by High-Speed Single-Molecule Microscopy" Viruses 13, no. 2: 167. https://doi.org/10.3390/v13020167
APA StyleJunod, S. L., Saredy, J., & Yang, W. (2021). Nuclear Import of Adeno-Associated Viruses Imaged by High-Speed Single-Molecule Microscopy. Viruses, 13(2), 167. https://doi.org/10.3390/v13020167