
Porcine Reproductive and Respiratory Syndrome (PRRS) Epidemiology in an Integrated Pig Company of Northern Italy: A Multilevel Threat Requiring Multilevel Interventions

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Preparation
2.2. Reconstruction of Viral Population Dynamics and Within-Company Spreading
2.3. Strain Migration among Integrated Pig Productive Chains
2.4. Continuous Phylogeographic Analysis
- 1.
- The viral lineages tendency to remain in and/or to disperse to areas associated with higher raster (i.e., environmental variable) values. For this purpose, two statistics were calculated: E (i.e., the mean of the environmental values extracted at the nodes’ position) and R (i.e., the proportion of branches for which the environmental value recorded at the oldest node position is higher than the environmental value recorded at the youngest node position). Therefore, E measures the tendency of tree nodes to remain located in lower/higher environmental values, while R measures the tendency of lineages to disperse towards lower/higher environmental values. To account for phylogenetic uncertainness, these two statistics were computed from a sample of 100 posterior trees, obtaining their posterior distribution, which was compared to a null distribution of the same metric computed after having randomized phylogenetic node positions within the study area, under the constraint that branch lengths, tree topology, and root position are unchanged. Thus, their statistical significance was assessed by BF calculation. For example, the BF associated with the statistic E was calculated as the posterior odds that Eestimated > Erandomised (if the environmental variable is considered to attract the lineages, the opposite Eestimated < Erandomised if repulsing them) divided by the equivalent prior odds (assuming the prior probability for Eestimated > Erandomised to be 0.5). The same approach was used for the R statistics.
- 2.
- The association between a particular environmental variable and the dispersal velocity of the considered strains. Two models of spatial movements were considered: (1) “straight line (SL) path” model, assuming a straight movement between the starting and ending locations of each branch (i.e., the branch weight is computed as the sum of raster cells covered by the straight line); (2) “least cost (LC) path” model, using a least-cost algorithm (i.e., the branch weight is computed as the sum of the values of cells transition values between neighboring cells along the least-cost path). In this model, the analyzed environmental variable can be considered both as a conductance (i.e., favoring viral dispersal through the cells with higher values) or resistance factor (i.e., allowing an easier dispersal through cells with lower values). Both instances were tested for each considered factor. The obtained “environmental” weights were used to calculate a regression with the branch duration, and the corresponding coefficient of determination (R2env) was obtained. A null coefficient of determination (R2null) was also calculated assuming a null raster (i.e., assuming that only the spatial distance of each movement affects the branch duration). The statistic Q = R2env − R2null was selected as the final outcome and describes how much the regression is strengthened when the spatial variation in the environmental variable is included. To account for the phylogenetic uncertainness, the Q statistic was calculated for a set of 100 trees sampled from the posterior distribution. A proportion of more than 90% of positive Q values > 0 was considered suggestive of environmental values effect and further tested through BF calculation. For this purpose, the same randomization procedure previously described was used and the BF for an environmental factor was approximated by the posterior odds that Qestimated > Qrandomised divided by the equivalent prior odds (still setting the prior probability for Qestimated > Qrandomised to 0.5).
3. Results
3.1. Dataset
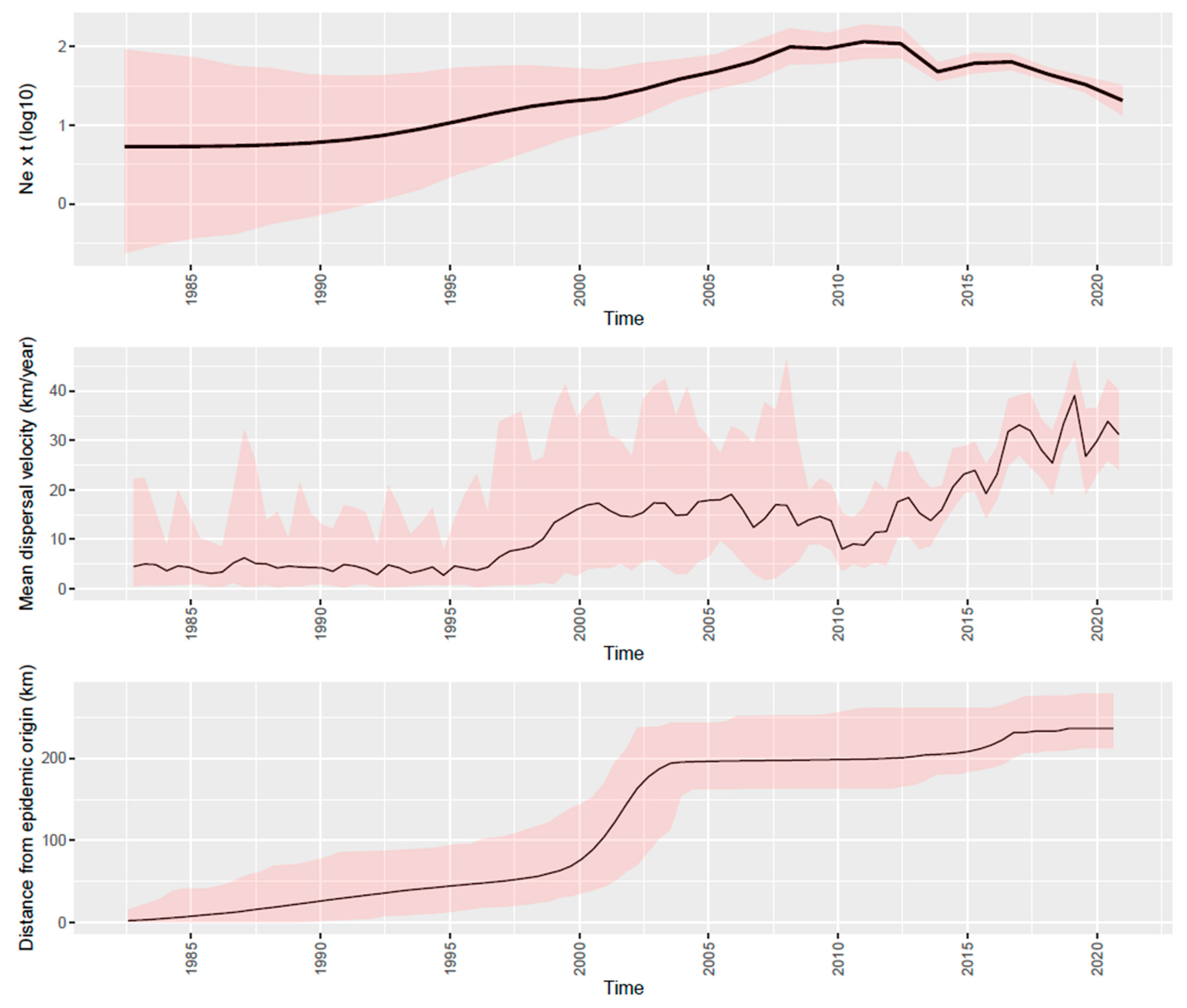
3.2. PRRSV Strain History, Evolution, and Population Dynamics
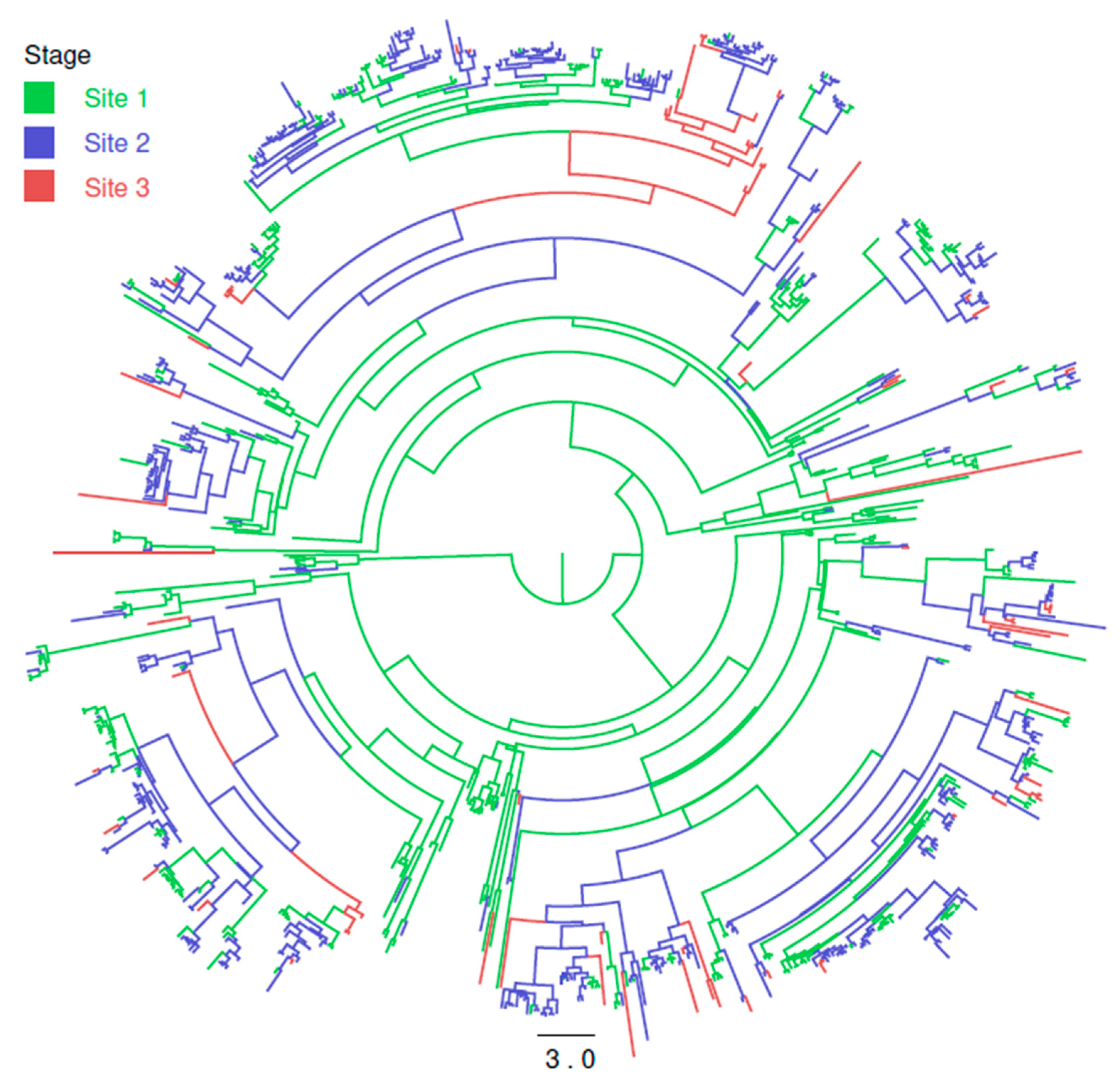
3.3. Within Integrated Pig Production Chain Strain Flow
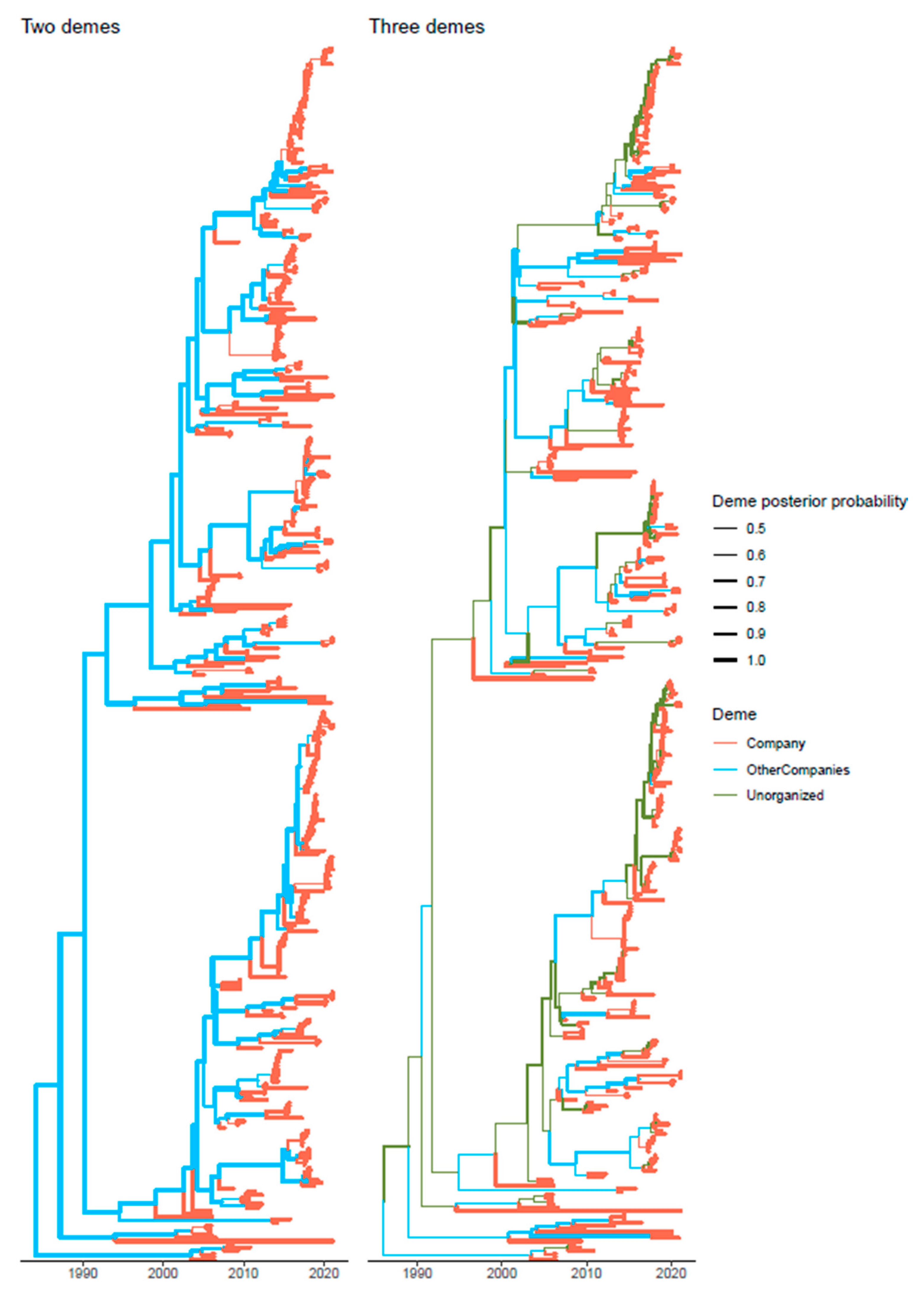
3.4. Strain Migration among Integrated Pig Production Chains
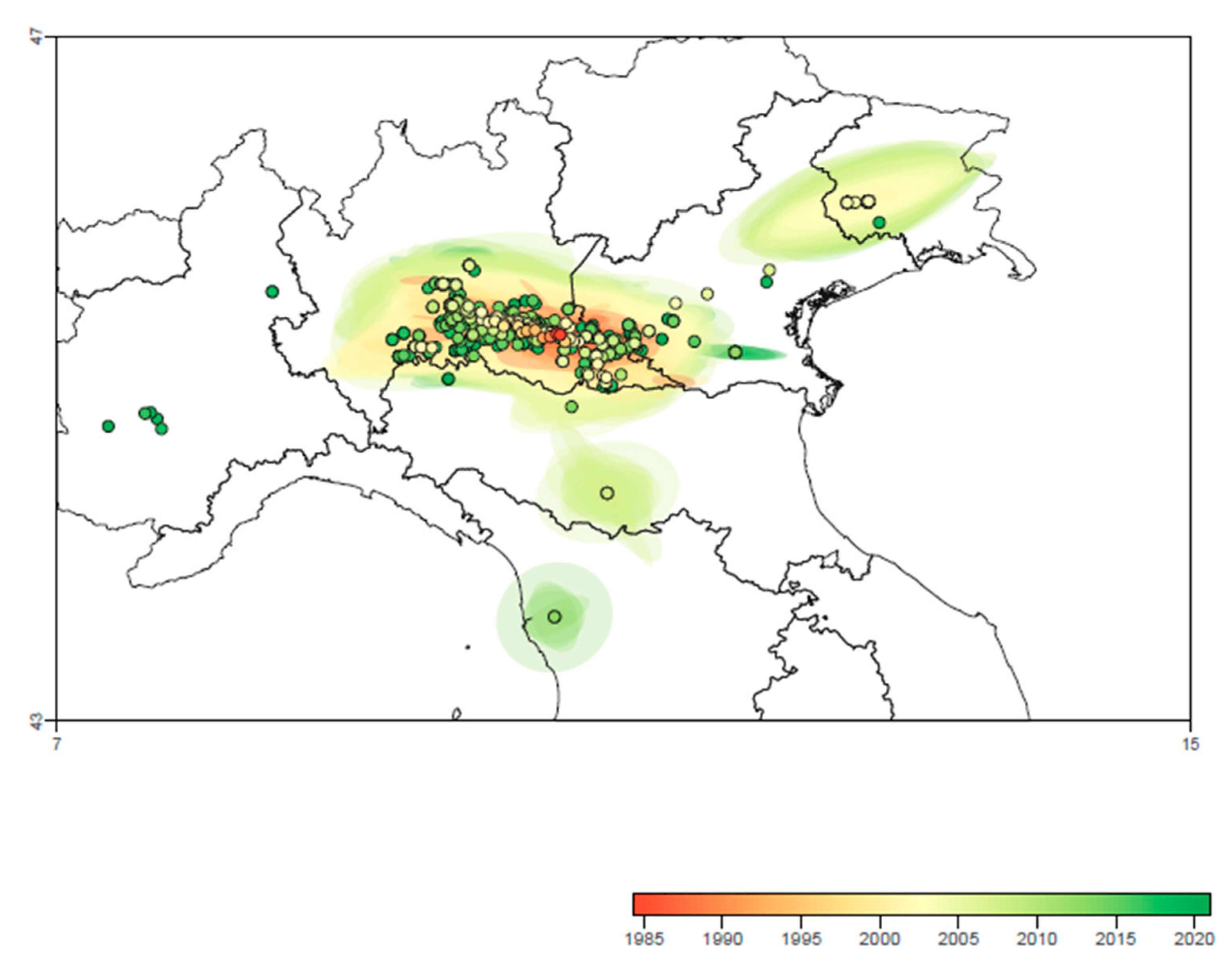
3.5. Continuous Phylogeography
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kappes, M.A.; Faaberg, K.S. PRRSV Structure, Replication and Recombination: Origin of Phenotype and Genotype Diversity. Virology 2015, 479–480, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S. Why Are RNA Virus Mutation Rates so Damn High? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of Evolutionary Change in Viruses: Patterns and Determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Canelli, E.; Catella, A.; Borghetti, P.; Ferrari, L.; Ogno, G.; de Angelis, E.; Corradi, A.; Passeri, B.; Bertani, V.; Sandri, G.; et al. Phenotypic Characterization of a Highly Pathogenic Italian Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Type 1 Subtype 1 Isolate in Experimentally Infected Pigs. Vet. Microbiol. 2017, 210, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, X.; Yu, H.; Jiang, Y.; Gao, F.; Tong, W.; Li, L.; Li, H.; Yang, S.; Chen, P.; et al. The Emergence of a Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus with Additional 120aa Deletion in Nsp2 Region in Jiangxi, China. Transbound. Emerg. Dis. 2018, 65, 1740–1748. [Google Scholar] [CrossRef]
- Ruedas-Torres, I.; Rodríguez-Gómez, I.M.; Sánchez-Carvajal, J.M.; Larenas-Muñoz, F.; Pallarés, F.J.; Carrasco, L.; Gómez-Laguna, J. The Jigsaw of PRRSV Virulence. Vet. Microbiol. 2021, 260, 109168. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.Y.; Hon, C.C.; Hui, R.K.H.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C.C. Molecular Epidemiology of PRRSV: A Phylogenetic Perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef]
- Stadejek, T.; Stankevicius, A.; Murtaugh, M.P.; Oleksiewicz, M.B. Molecular Evolution of PRRSV in Europe: Current State of Play. Vet. Microbiol. 2013, 165, 21–28. [Google Scholar] [CrossRef]
- Franzo, G.; Dotto, G.; Cecchinato, M.; Pasotto, D.; Martini, M.; Drigo, M.; Franzo, G. Phylodynamic Analysis of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) in Italy: Action of Selective Pressures and Interactions between Different Clades. Infect. Genet. Evol. 2015, 31, 149–157. [Google Scholar] [CrossRef]
- Corzo, C.A.; Mondaca, E.; Wayne, S.; Torremorell, M.; Dee, S.; Davies, P.; Morrison, R.B. Control and Elimination of Porcine Reproductive and Respiratory Syndrome Virus. Virus Res. 2010, 154, 185–192. [Google Scholar] [CrossRef]
- Nieuwenhuis, N.; Duinhof, T.F.; Nes, A. van Economic Analysis of Outbreaks of Porcine Reproductive and Respiratory Syndrome Virus in Nine Sow Herds. Vet. Rec. 2012, 170, 225. [Google Scholar] [CrossRef]
- Rowland, R.R.R.; Morrison, R.B. Challenges and Opportunities for the Control and Elimination of Porcine Reproductive and Respiratory Syndrome Virus. Transbound. Emerg. Dis. 2012, 59, 55–59. [Google Scholar] [CrossRef]
- Chae, C. Commercial Prrs Modified-live Virus Vaccines. Vaccines 2021, 9, 185. [Google Scholar] [CrossRef]
- Cho, J.G.; Dee, S.A. Porcine Reproductive and Respiratory Syndrome Virus. Theriogenology 2006, 66, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Candotti, P.; Rota-Nodari, S.; Leotti, G.; Longo, S.; Joisel, F. Relationship between Piglet PRRS Seroprevalence at the End of Nursery and Reproduction Disorders Due to PRRS in Sows. In Proceedings of the 19th IPVS Congress, Copenhagen, Denmark, 16–19 July 2006; Volume 1, p. 54. [Google Scholar]
- Stadejek, T.; Oleksiewicz, M.B.; Scherbakov, A.V.; Timina, A.M.; Krabbe, J.S.; Chabros, K.; Potapchuk, D. Definition of Subtypes in the European Genotype of Porcine Reproductive and Respiratory Syndrome Virus: Nucleocapsid Characteristics and Geographical Distribution in Europe. Arch. Virol. 2008, 153, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Alkhamis, M.A.; Arruda, A.G.; Morrison, R.B.; Perez, A.M. Novel Approaches for Spatial and Molecular Surveillance of Porcine Reproductive and Respiratory Syndrome Virus (PRRSv) in the United States. Sci. Rep. 2017, 7, 4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makau, D.N.; Alkhamis, M.A.; Paploski, I.a.D.; Corzo, C.A.; Lycett, S.; VanderWaal, K. Integrating Animal Movements with Phylogeography to Model the Spread of PRRSV in the USA. Virus Evol. 2021, 7, 1–11. [Google Scholar] [CrossRef]
- Drigo, M.; Franzo, G.; Gigli, A.; Martini, M.; Mondin, A.; Gracieux, P.; Ceglie, L. The Impact of Porcine Reproductive and Respiratory Syndrome Virus Genetic Heterogeneity on Molecular Assay Performances. J. Virol. Methods 2014, 202, 79–86. [Google Scholar] [CrossRef]
- Standley, K. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. (Outlines Version 7). Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple Alignment of Nucleotide Sequences Guided by Amino Acid Translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A Genetic Algorithm for Recombination Detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nature Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestet, C. Ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A (Stat. Soc.) 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees With Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- de Maio, N.; Wu, C.H.; O’Reilly, K.M.; Wilson, D. New Routes to Phylogeography: A Bayesian Structured Coalescent Approximation. PLoS Genet. 2015, 11, e1005421. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; de Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, T.G.; Kühnert, D.; Popinga, A.; Welch, D.; Drummond, A.J. Efficient Bayesian Inference under the Structured Coalescent. Bioinformatics 2014, 30, 2272–2279. [Google Scholar] [CrossRef] [Green Version]
- Ewing, G.; Rodrigo, A. Estimating Population Parameters Using the Structured Serial Coalescent with Bayesian MCMC Inference When Some Demes Are Hidden. Evol. Bioinform. 2006, 2, 117693430600200. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography Takes a Relaxed Random Walk in Continuous Space and Time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Dellicour, S.; Rose, R.; Pybus, O.G. Explaining the Geographic Spread of Emerging Epidemics: A Framework for Comparing Viral Phylogenies and Environmental Landscape Data. BMC Bioinform. 2016, 17, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellicour, S.; Rose, R.; Faria, N.R.; Lemey, P.; Pybus, O.G. SERAPHIM: Studying Environmental Rasters and Phylogenetically Informed Movements. Bioinformatics 2016, 32, 3204–3206. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Cecchinato, M.; Martini, M.; Ceglie, L.; Gigli, A.; Drigo, M. Observation of High Recombination Occurrence of Porcine Reproductive and Respiratory Syndrome Virus in Field Condition. Virus Res. 2014, 194, 159–166. [Google Scholar] [CrossRef]
- Holtkamp, D.J.; Yeske, P.E.; Polson, D.D.; Melody, J.L.; Philips, R.C. A Prospective Study Evaluating Duration of Swine Breeding Herd PRRS Virus-Free Status and Its Relationship with Measured Risk. Prev. Vet. Med. 2010, 96, 186–193. [Google Scholar] [CrossRef]
- Yoon, S.H.; Kim, H.; Park, B.; Kim, H. Tracing the Genetic History of Porcine Reproductive and Respiratory Syndrome Viruses Derived from the Complete ORF 5-7 Sequences: A Bayesian Coalescent Approach. Arch. Virol. 2012, 157, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.G.; Vilalta, C.; Perez, A.; Morrison, R. Land Altitude, Slope, and Coverage as Risk Factors for Porcine Reproductive and Respiratory Syndrome (PRRS) Outbreaks in the United States. PLoS ONE 2017, 12, e0172638. [Google Scholar] [CrossRef] [Green Version]
- Jara, M.; Rasmussen, D.A.; Corzo, C.A.; Machado, G. Porcine Reproductive and Respiratory Syndrome Virus Dissemination across Pig Production Systems in the United States. Transbound. Emerg. Dis. 2021, 68, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Pesente, P.; Rebonato, V.; Sandri, G.; Giovanardi, D.; Ruffoni, L.S.; Torriani, S. Phylogenetic Analysis of ORF5 and ORF7 Sequences of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) from PRRS-Positive Italian Farms: A Showcase for PRRSV Epidemiology and Its Consequences on Farm Management. Vet. Microbiol. 2006, 114, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Velthuis, A.G.J.; Mourits, M.C.M. Effectiveness of Movement-Prevention Regulations to Reduce the Spread of Foot-and-Mouth Disease in The Netherlands. Prev. Vet. Med. 2007, 82, 262–281. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, B.; Besser, T.E.; Gay, J.M.; Fox, L.K.; Davis, M.A.; Cobbold, R.N.; Berge, A.C.B.; Hancock, D.D. The Role of Animal Movement, Including off-Farm Rearing of Heifers, in the Interherd Transmission of Multidrug-Resistant Salmonella. J. Dairy Sci. 2009, 92, 4229–4238. [Google Scholar] [CrossRef]
- Machado, G.; Vilalta, C.; Recamonde-Mendoza, M.; Corzo, C.; Torremorell, M.; Perez, A.; VanderWaal, K. Identifying Outbreaks of Porcine Epidemic Diarrhea Virus through Animal Movements and Spatial Neighborhoods. Sci. Rep. 2019, 9, 457. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Tucciarone, C.M.; Moreno, A.; Legnardi, M.; Massi, P.; Tosi, G.; Trogu, T.; Ceruti, R.; Pesente, P.; Ortali, G.; et al. Phylodynamic Analysis and Evaluation of the Balance between Anthropic and Environmental Factors Affecting IBV Spreading among Italian Poultry Farms. Sci. Rep. 2020, 10, 7289. [Google Scholar] [CrossRef]
- Otake, S.; Dee, S.; Corzo, C.; Oliveira, S.; Deen, J. Long-Distance Airborne Transport of Infectious PRRSV and Mycoplasma Hyopneumoniae from a Swine Population Infected with Multiple Viral Variants. Vet. Microbiol. 2010, 145, 198–208. [Google Scholar] [CrossRef]
- Pitkin, A.; Deen, J.; Dee, S. Use of a Production Region Model to Assess the Airborne Spread of Porcine Reproductive and Respiratory Syndrome Virus. Vet. Microbiol. 2009, 136, 1–7. [Google Scholar] [CrossRef]
- Perez, A.M.; Linhares, D.C.L.; Arruda, A.G.; VanderWaal, K.; Machado, G.; Vilalta, C.; Sanhueza, J.M.; Torrison, J.; Torremorell, M.; Corzo, C.A. Individual or Common Good? Voluntary Data Sharing to Inform Disease Surveillance Systems in Food Animals. Front. Vet. Sci. 2019, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.; Hoff, S.; Muñoz-Zanzi, C.; Yoon, K.J.; Roof, M.; Burkhardt, A.; Zimmerman, J. Effect of Temperature and Relative Humidity on the Stability of Infectious Porcine Reproductive and Respiratory Syndrome Virus in Aerosols. Vet. Res. 2007, 38, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dee, S.; Otake, S.; Oliveira, S.; Deen, J. Evidence of Long Distance Airborne Transport of Porcine Reproductive and Respiratory Syndrome Virus and Mycoplasma Hyopneumoniae. Vet. Res. 2009, 40, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzo, G.; Barbierato, G.; Pesente, P.; Legnardi, M.; Tucciarone, C.M.; Sandri, G.; Drigo, M. Porcine Reproductive and Respiratory Syndrome (PRRS) Epidemiology in an Integrated Pig Company of Northern Italy: A Multilevel Threat Requiring Multilevel Interventions. Viruses 2021, 13, 2510. https://doi.org/10.3390/v13122510
Franzo G, Barbierato G, Pesente P, Legnardi M, Tucciarone CM, Sandri G, Drigo M. Porcine Reproductive and Respiratory Syndrome (PRRS) Epidemiology in an Integrated Pig Company of Northern Italy: A Multilevel Threat Requiring Multilevel Interventions. Viruses. 2021; 13(12):2510. https://doi.org/10.3390/v13122510
Chicago/Turabian StyleFranzo, Giovanni, Giacomo Barbierato, Patrizia Pesente, Matteo Legnardi, Claudia Maria Tucciarone, Giampietro Sandri, and Michele Drigo. 2021. "Porcine Reproductive and Respiratory Syndrome (PRRS) Epidemiology in an Integrated Pig Company of Northern Italy: A Multilevel Threat Requiring Multilevel Interventions" Viruses 13, no. 12: 2510. https://doi.org/10.3390/v13122510
APA StyleFranzo, G., Barbierato, G., Pesente, P., Legnardi, M., Tucciarone, C. M., Sandri, G., & Drigo, M. (2021). Porcine Reproductive and Respiratory Syndrome (PRRS) Epidemiology in an Integrated Pig Company of Northern Italy: A Multilevel Threat Requiring Multilevel Interventions. Viruses, 13(12), 2510. https://doi.org/10.3390/v13122510