
The Promotion of Genomic Instability in Human Fibroblasts by Adenovirus 12 Early Region 1B 55K Protein in the Absence of Viral Infection

 , , , and
, , , and 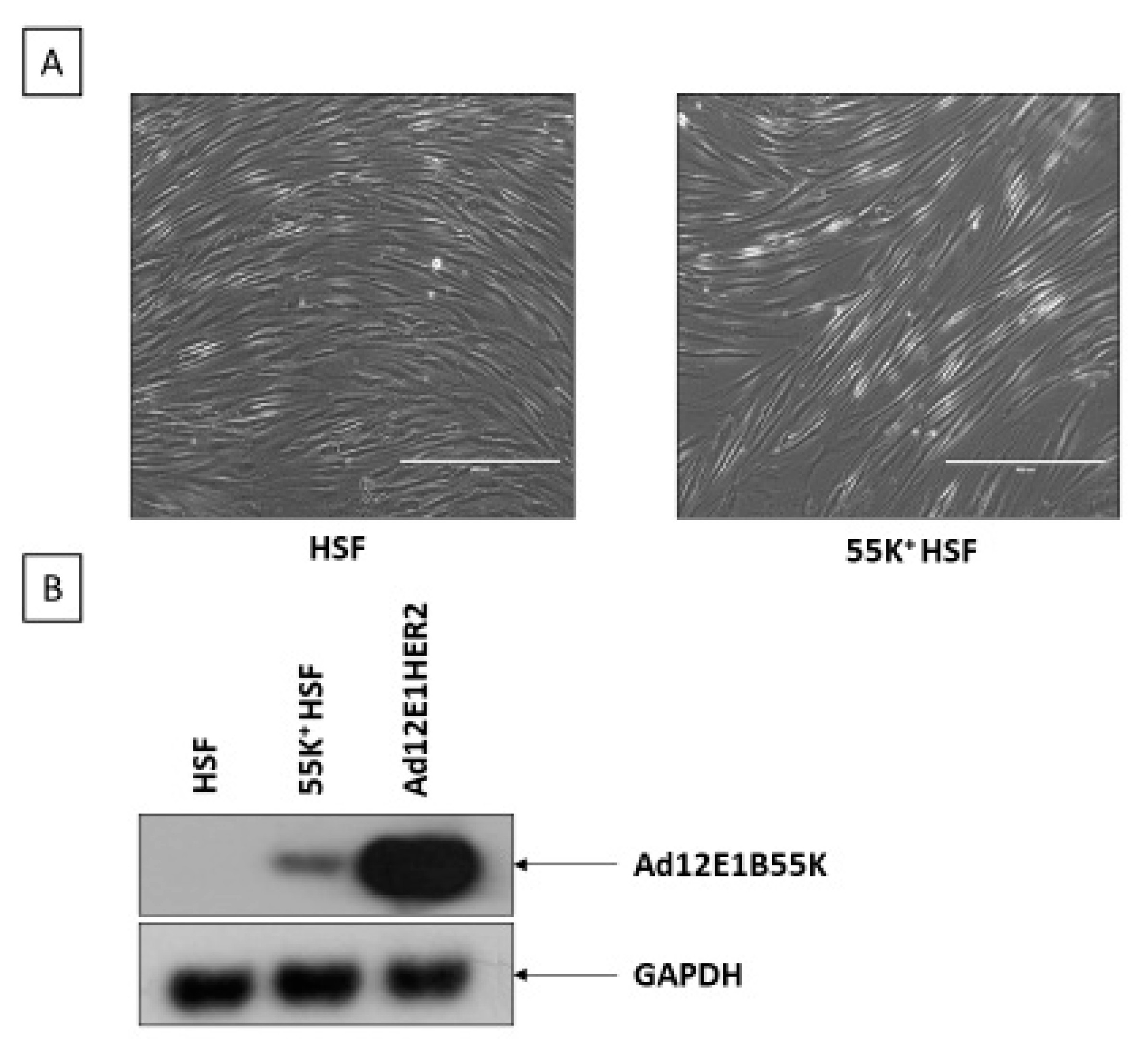{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Drug Treatments
2.2. Western Blotting and Antibodies
2.3. Co-Immunoprecipitation
2.4. Chromatin Preparation
2.5. Metaphase Spreads
2.6. Immunofluorescence Microscopy
2.7. Estimation of R-Loops
2.8. DNA Fiber Analysis
2.9. Statistical Analysis
3. Results
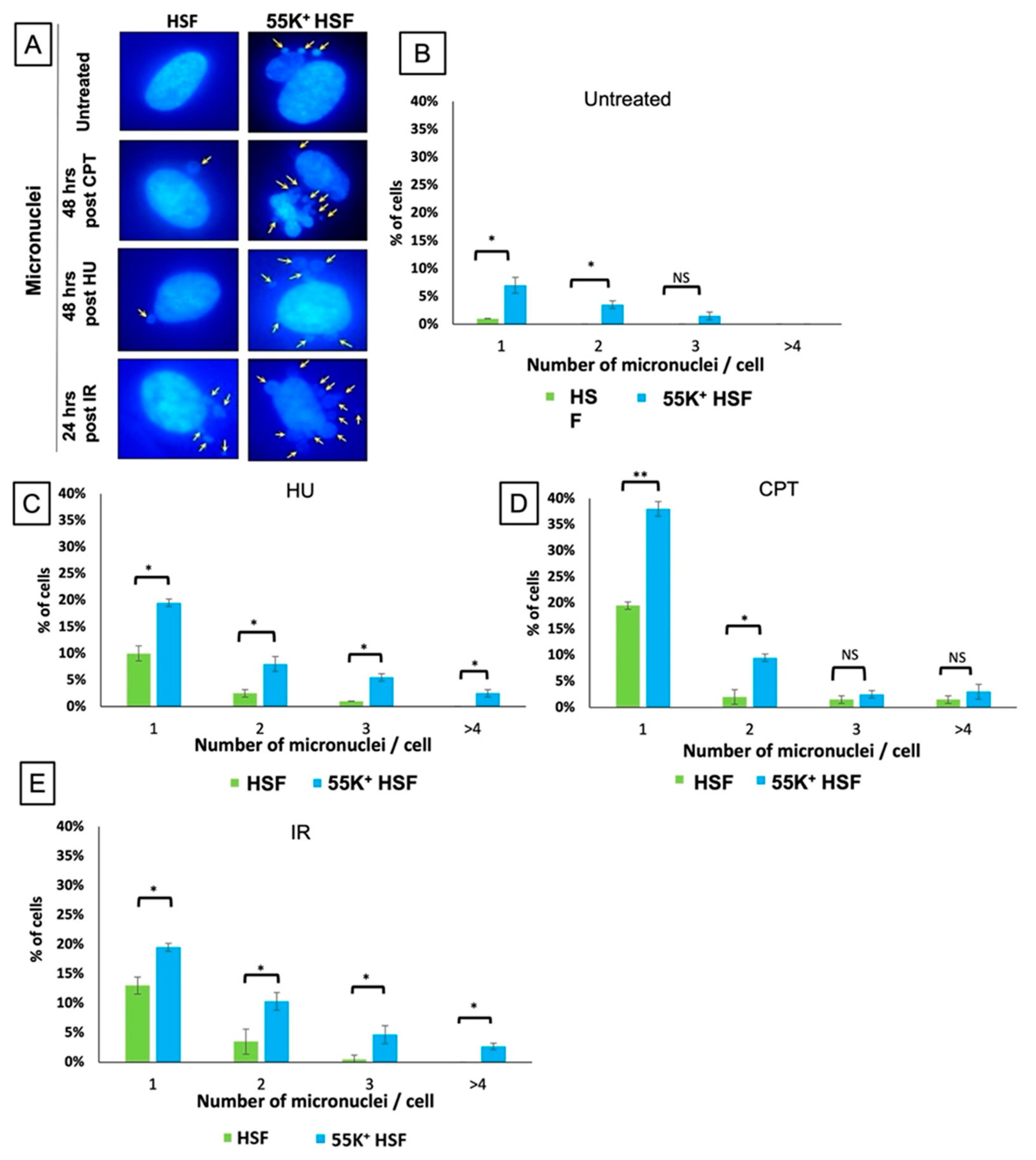
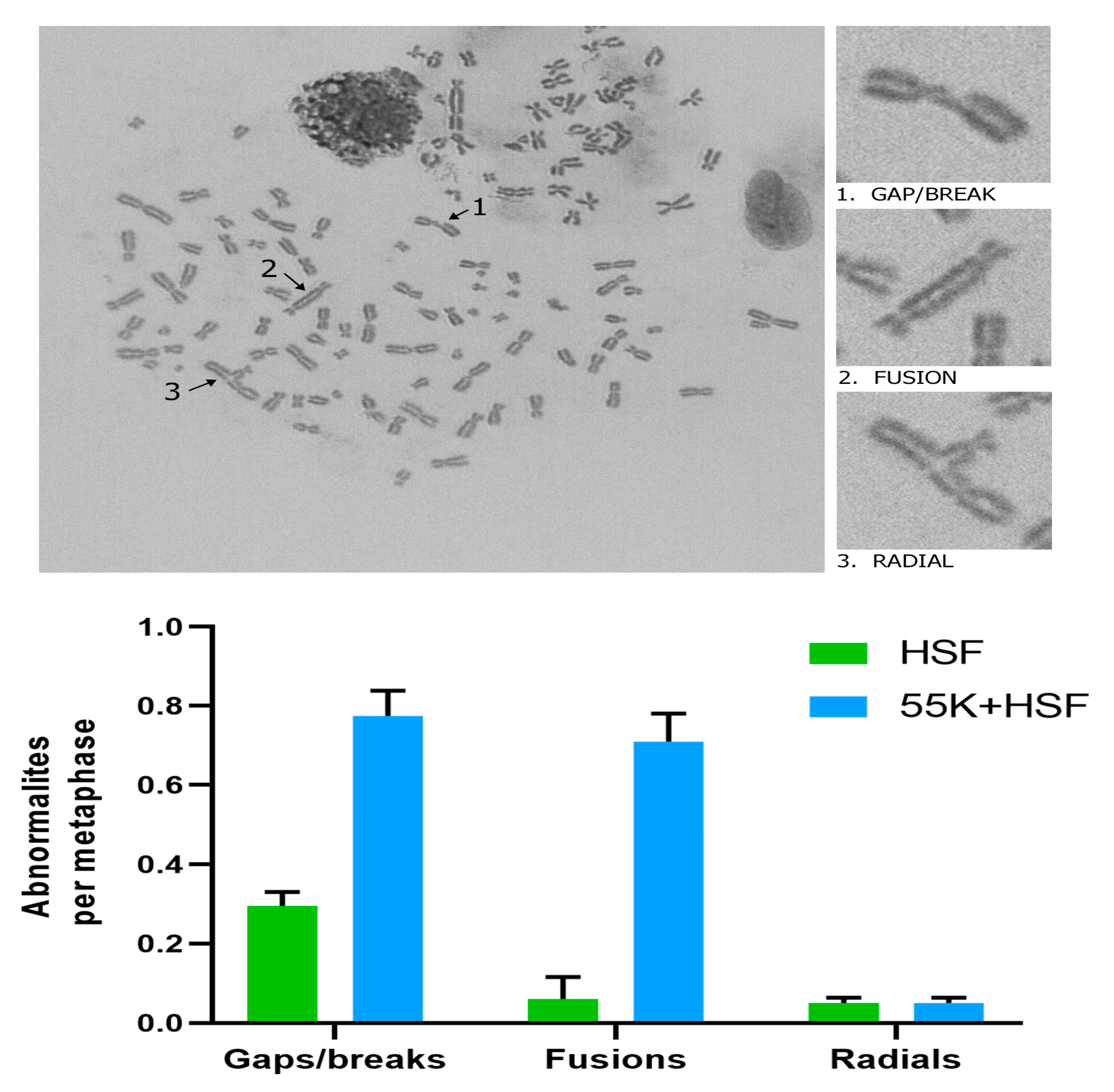
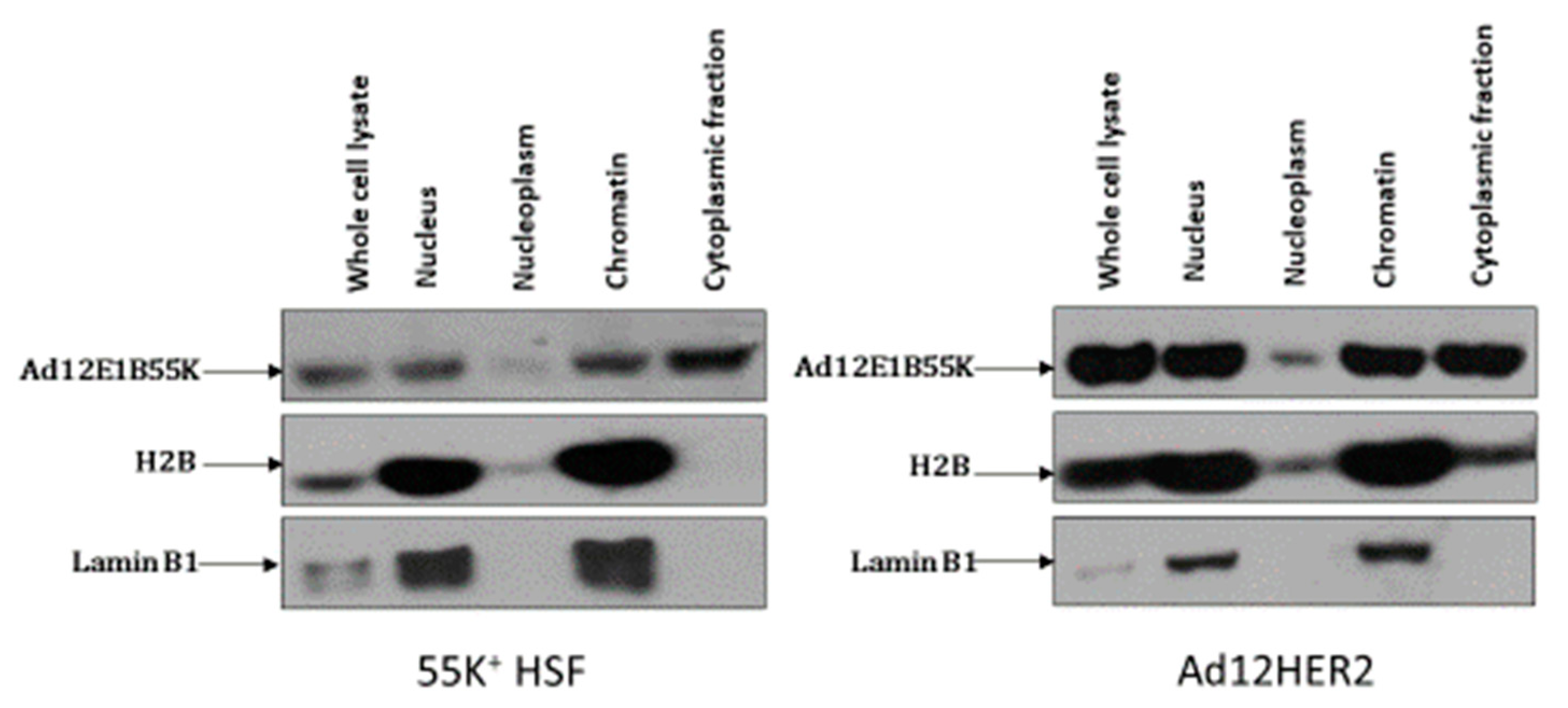
3.1. Ad12E1B55K Induces Genomic Instability in Human Skin Fibroblasts
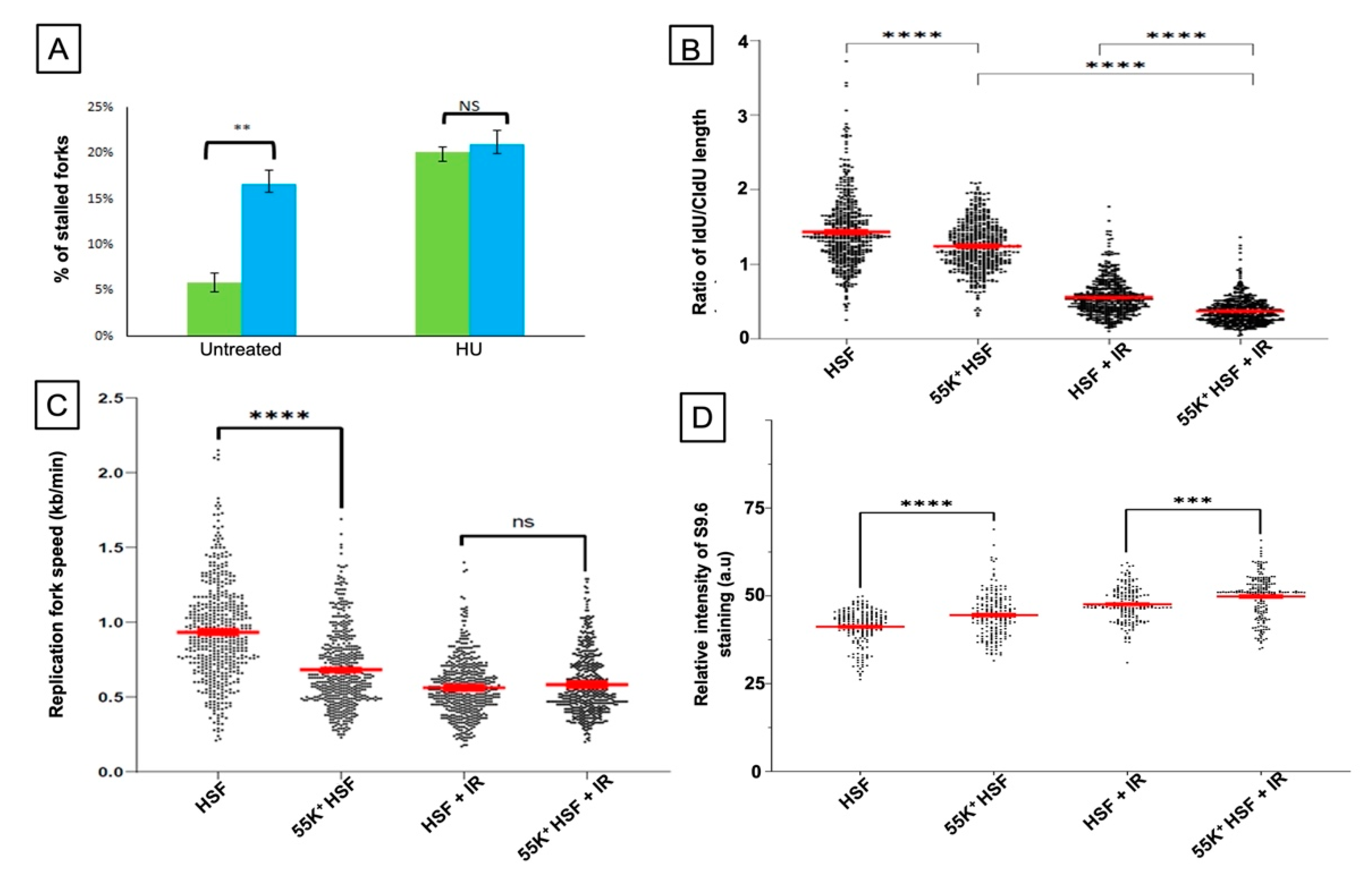
3.2. Ad12E1B55K Affects DNA Replication in HSFs and Leads to an Increase in R-Loop Formation
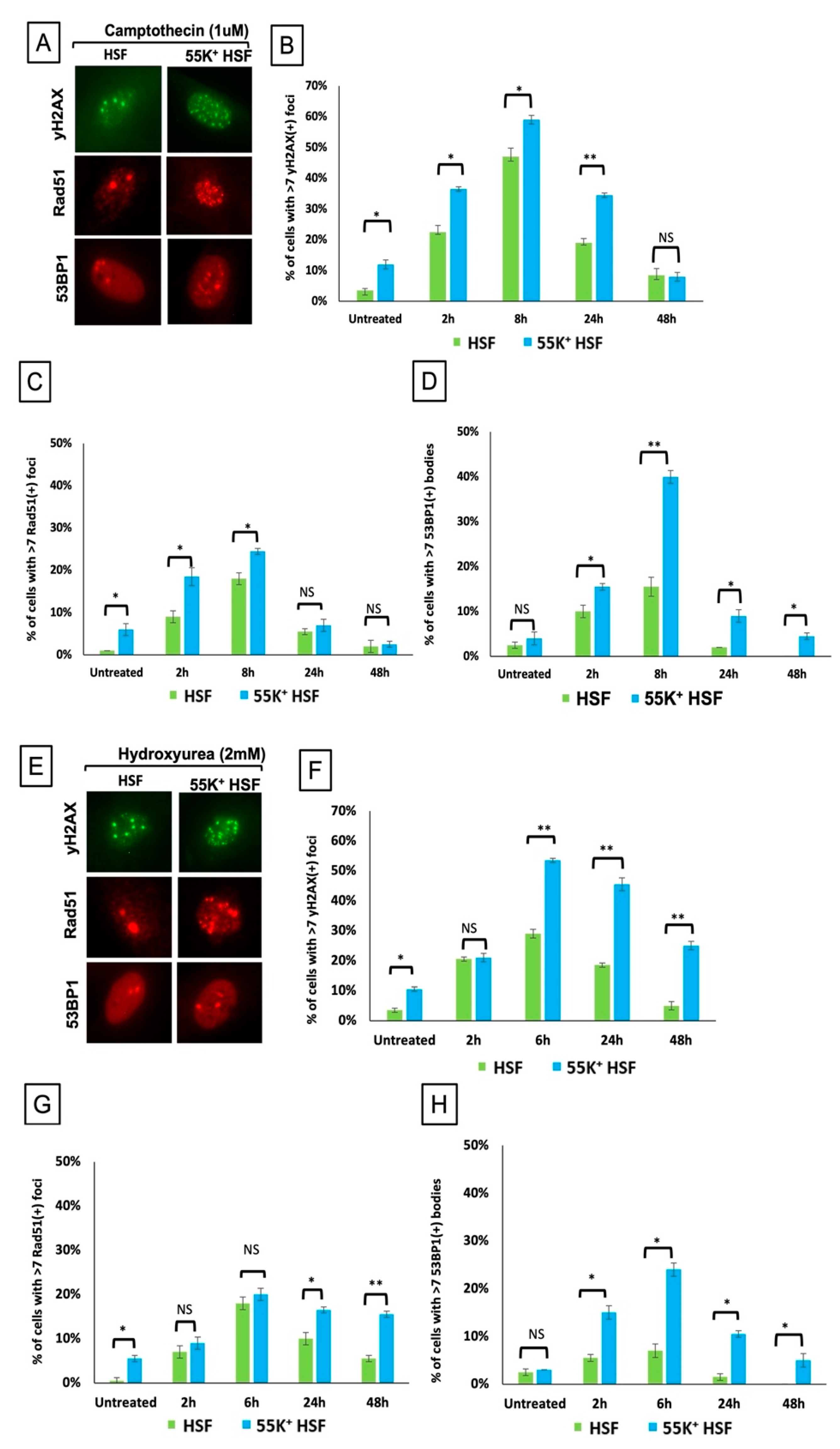
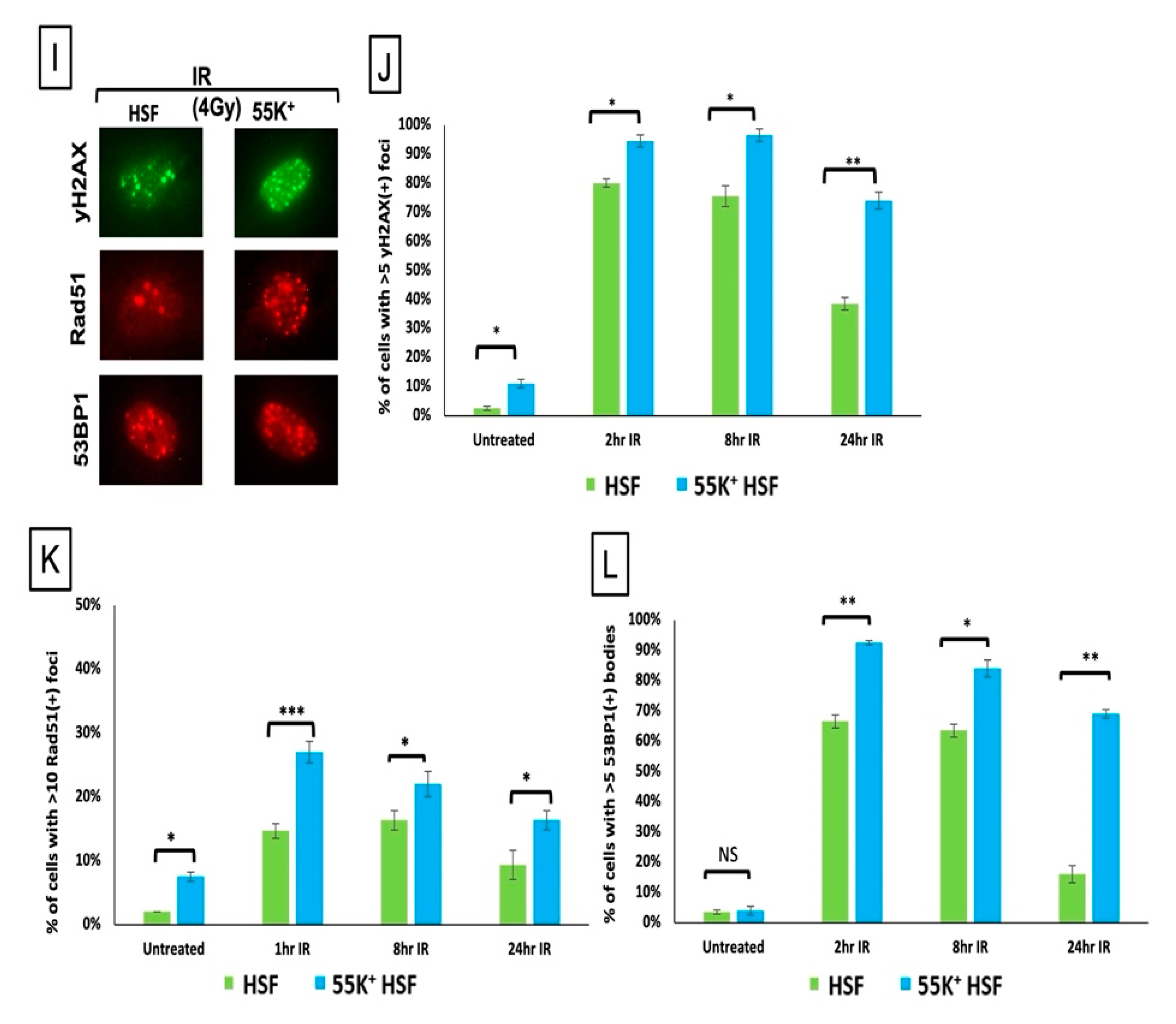
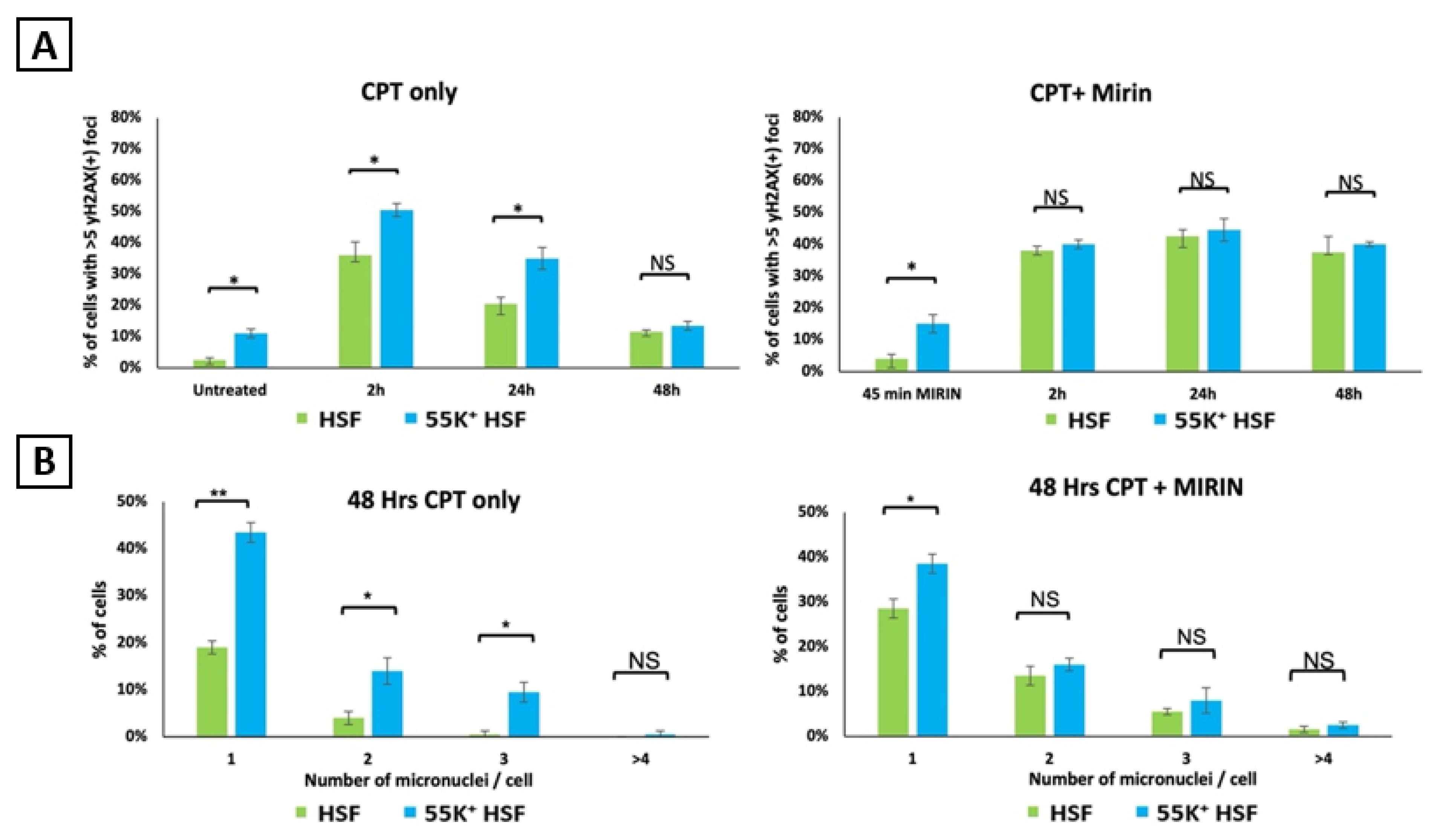
3.3. Ad12E1B55K Protein Sensitises HSFs to DNA Damaging Agents
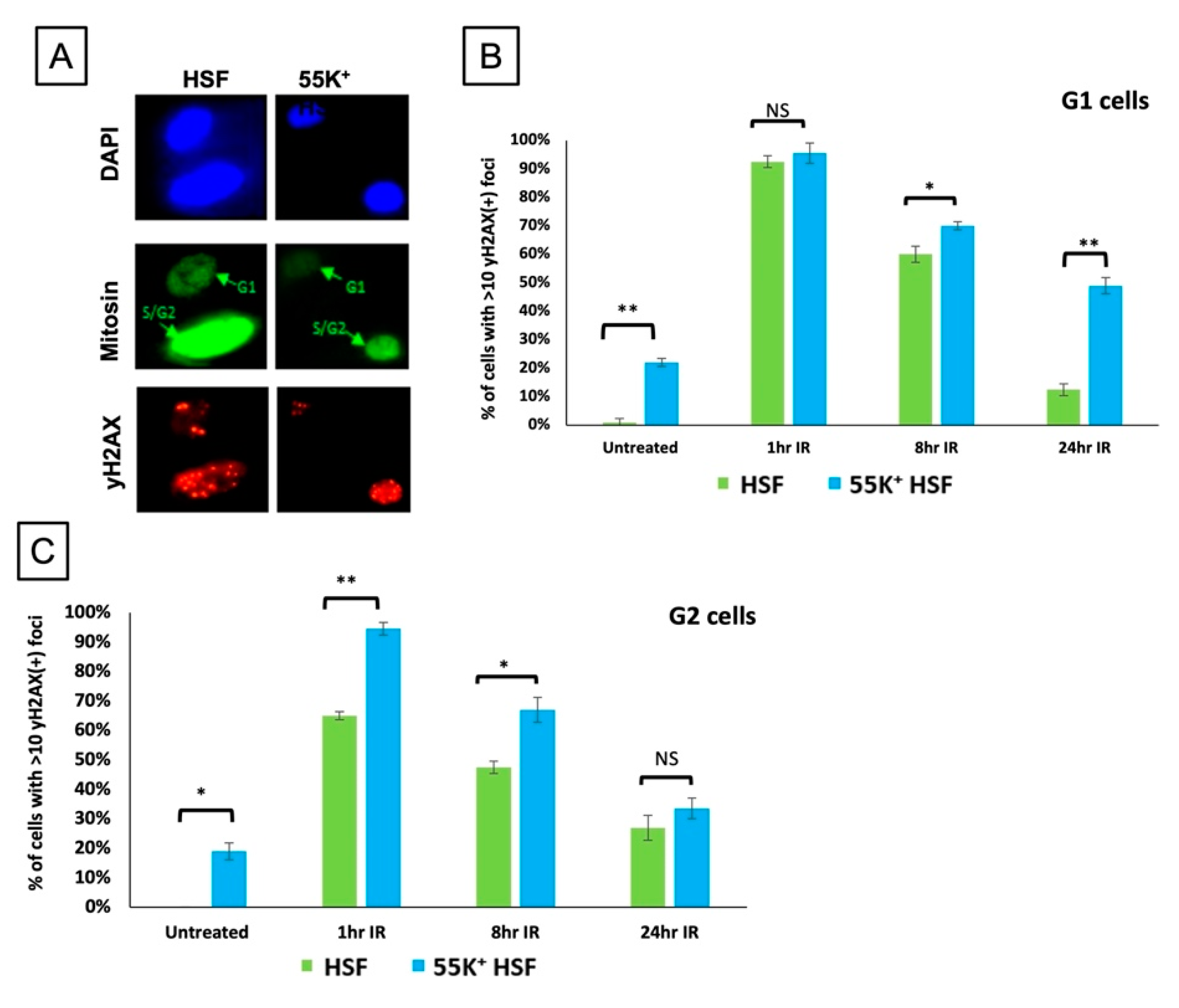
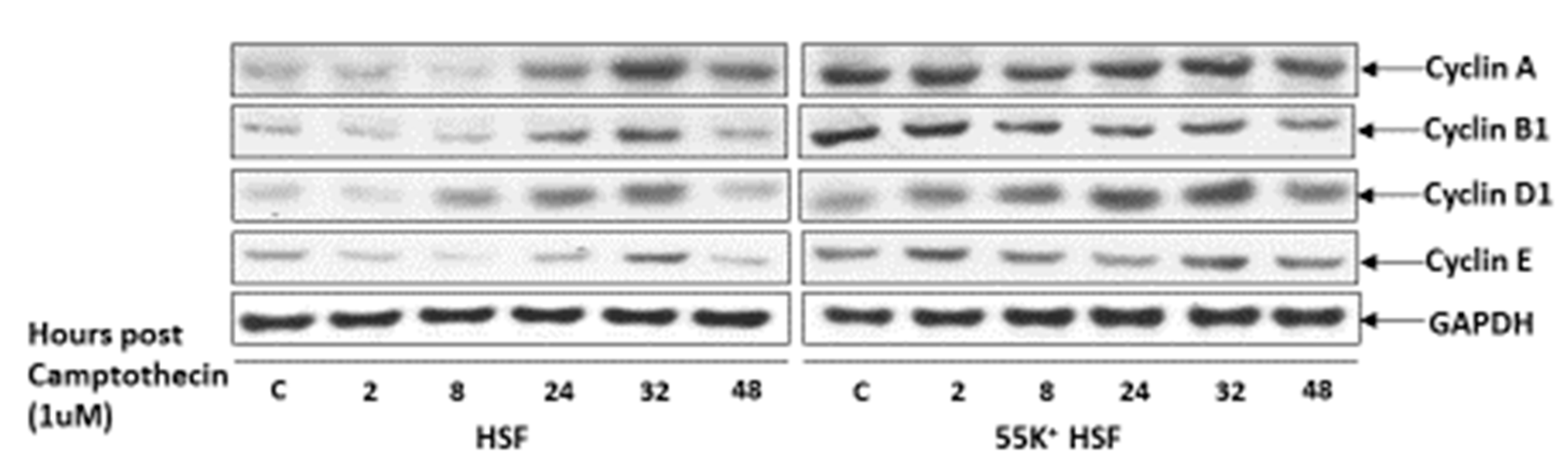
3.4. Ad12E1B55K Affects the Ability of HSFs to Undergo Cell Cycle Arrest following DNA Damage
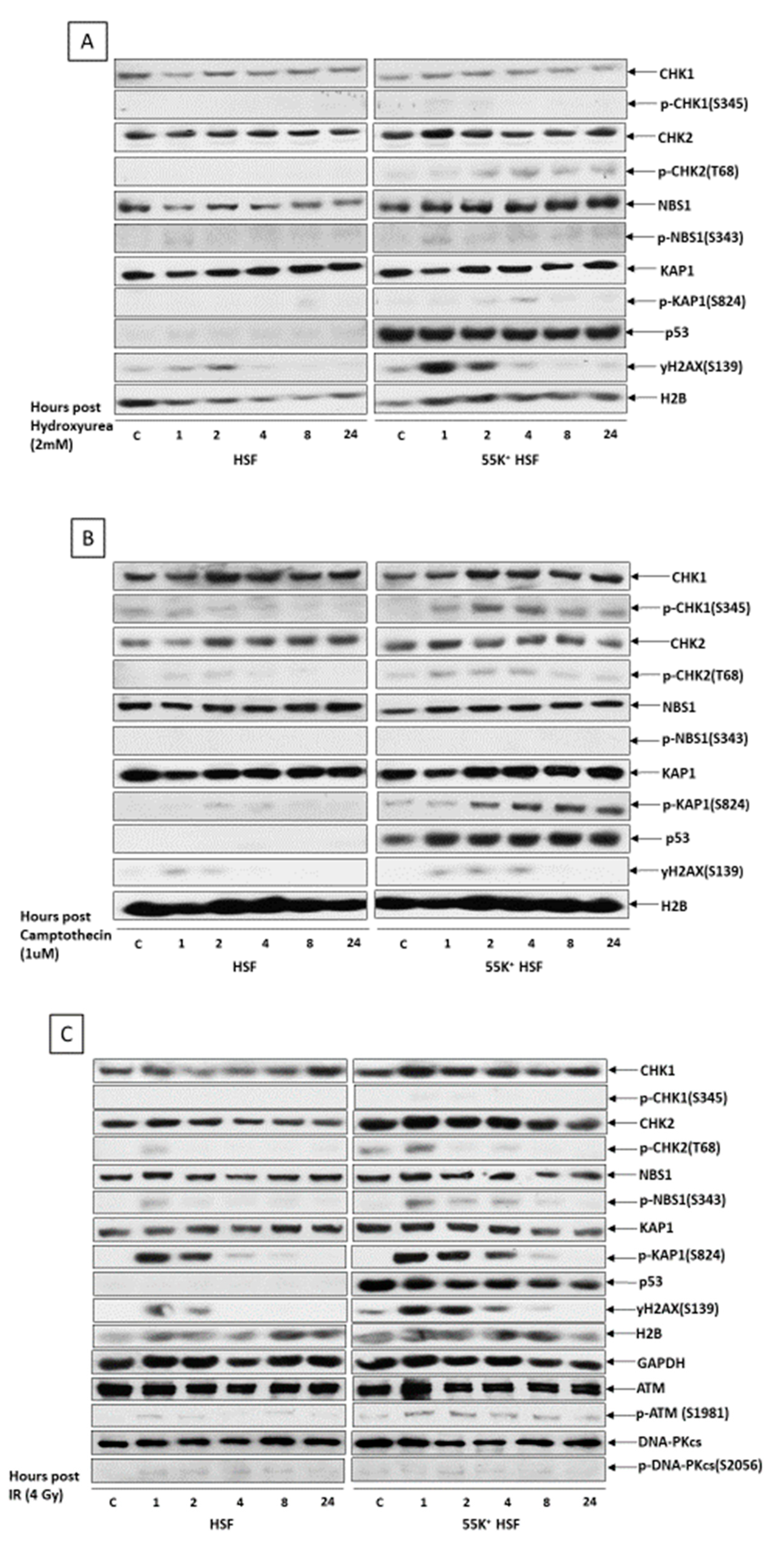
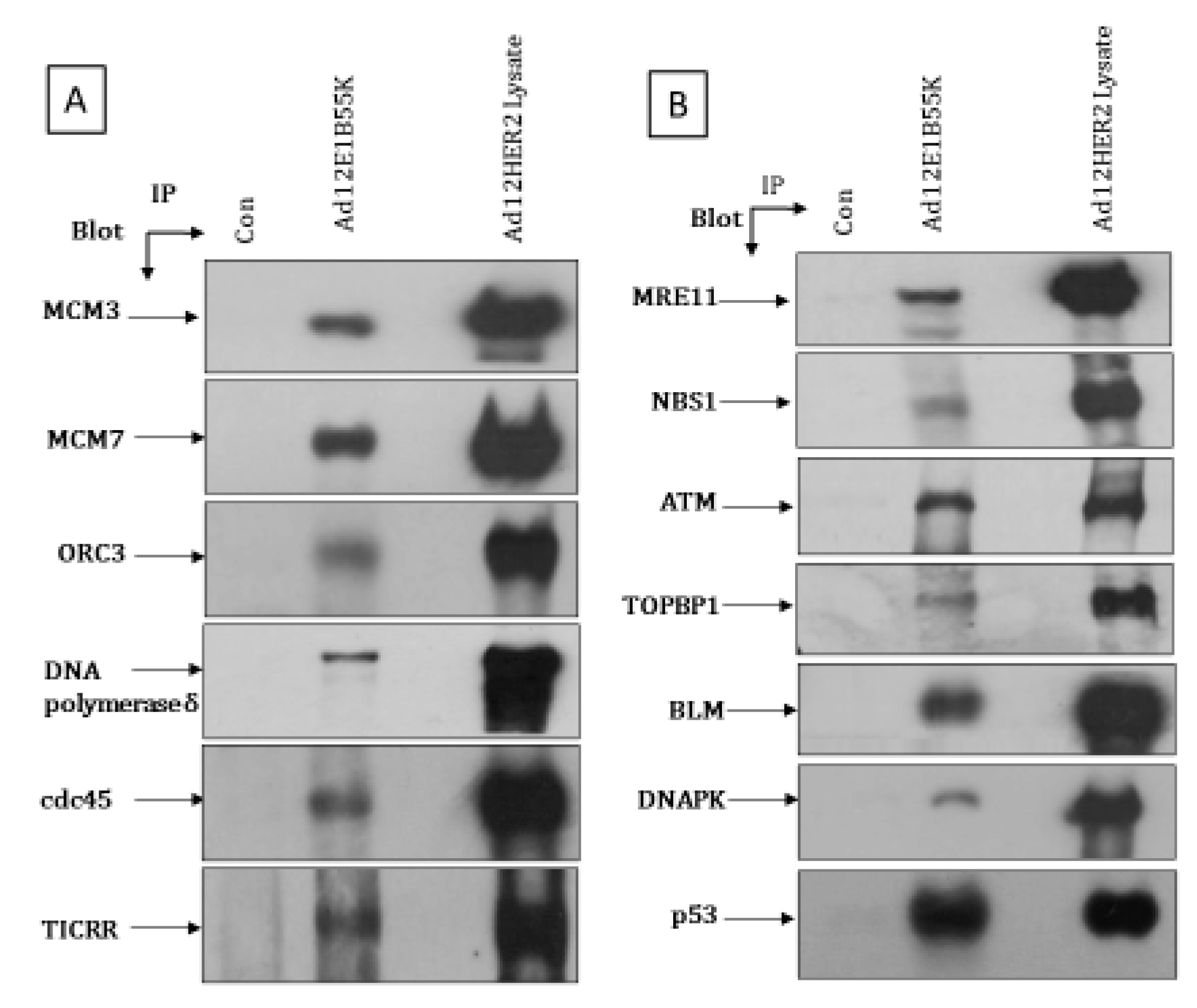
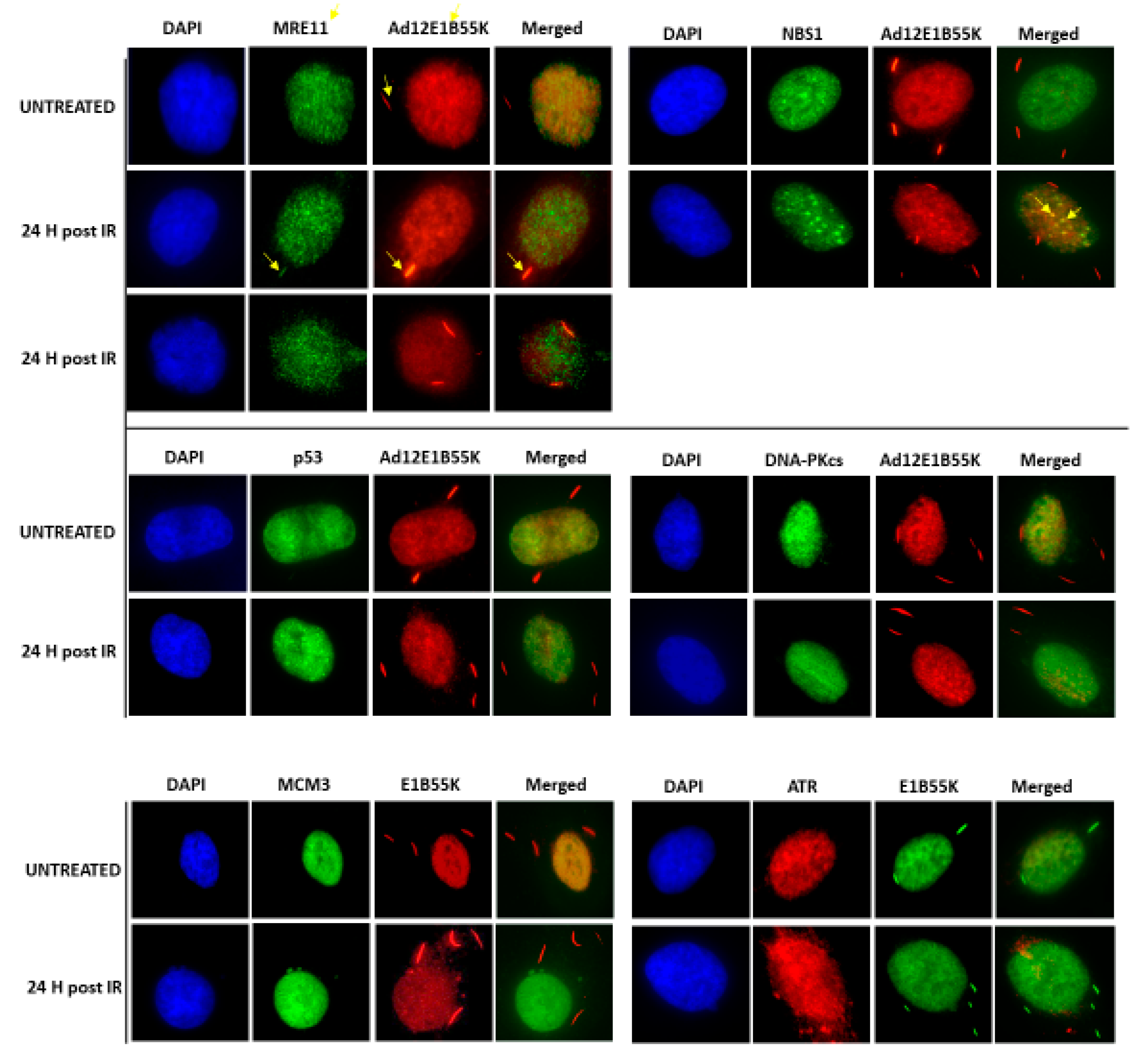
3.5. Association of Ad12E1B55K with Replication Machinery and DDR Pathway Components
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kajon, A.E.; Lynch, J.P. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Khanal, S.; Ghimire, P.; Dhamoon, A.S. The Repertoire of Adenovirus in Human Disease: The Innocuous to the Deadly. Biomedicines 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Huebner, R.J.; Rowe, W.P.; Turner, H.C.; Lane, W.T. Specific adenovirus complement-fixing antigens in virus-free hamster and rat tumors. Proc. Natl. Acad. Sci. USA 1963, 50, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Yabe, Y.; Trentin, J.J.; Taylor, G. Cancer Induction in Hamsters by Human Type 12 Adenovirus. Effect of Age and of Virus Dose. Exp. Biol. Med. 1962, 111, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Trentin, J.J.; Yabe, Y.; Taylor, G. The Quest for Human Cancer Viruses. Science 1962, 137, 835–841. [Google Scholar] [CrossRef]
- Freeman, A.E.; Black, P.H.; Wolford, R.; Huebner, R.J. Adenovirus type 12-rat embryo transformation system. J. Virol. 1967, 1, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.E.; Black, P.H.; Vanderpool, E.A.; Henry, P.H.; Austin, J.B.; Huebner, R.J. Transformation of primary rat embryo cells by adenovirus type 2. Proc. Natl. Acad. Sci. USA 1967, 58, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, P.H. Tumour Production in Immunosuppressed Rats with Cells Transformed in vitro by Adenovirus Type 2. J. Gen. Virol. 1972, 16, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, P.H.; McDougall, J.K.; Chen, L.B. In vitro traits of adenovirus-transformed cell lines and their relevance to tumorigenicity in nude mice. Cell 1977, 10, 669–678. [Google Scholar] [CrossRef]
- Mukai, N.; Kalter, S.S.; Cummins, L.B.; Matthews, V.A.; Nishida, T.; Nakajima, T. Retinal Tumor Induced in the Baboon by Human Adenovirus 12. Science 1980, 210, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Hausen, H.Z. Induction of Specific Chromosomal Aberrations by Adenovirus Type 12 in Human Embryonic Kidney Cells. J. Virol. 1967, 1, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- McDougall, J.K. Effects of Adenoviruses on the Chromosomes of Normal Human Cells and Cells Trisomic for an E Chromosome. Nature 1970, 225, 456–458. [Google Scholar] [CrossRef]
- McDougall, J.K. Adenovirus-induced Chromosome Aberrations in Human Cells. J. Gen. Virol. 1971, 12, 43–51. [Google Scholar] [CrossRef]
- Stich, H.; Van Hoosier, G.; Trentin, J. Viruses and mammalian chromosomes chromosome aberrations by human adenovirus type 12. Exp. Cell Res. 1964, 34, 400–403. [Google Scholar] [CrossRef]
- McDougall, J.K.; Vause, K.E.; Gallimore, P.H.; Dunn, A.R. Cytogenetic studies in permissive and abortive infections by adenovirus type 12. Int. J. Cancer 1974, 14, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, V.; Ares, M.; Weiner, A.M.; Francke, U. Human genes for U2 small nuclear RNA map to a major adenovirus 12 modification site on chromosome 17. Nature 1985, 314, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Durnam, D.M.; Menninger, J.C.; Chandler, S.H.; Smith, P.P.; McDougall, J.K. A fragile site in the human U2 small nuclear RNA gene cluster is revealed by adenovirus type 12 infection. Mol. Cell Biol. 1988, 8, 1863–1867. [Google Scholar] [PubMed]
- Bailey, A.D.; Li, Z.; Pavelitz, T.; Weiner, A.M. Adenovirus type 12-induced fragility of the human RNU2 locus requires U2 small nuclear RNA transcriptional regulatory elements. Mol. Cell. Biol. 1995, 15, 6246–6255. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Tomanin, R.; Smiley, J.R.; Bacchetti, S. Generation of a new adenovirus type 12-inducible fragile site by insertion of an artificial U2 locus in the human genome. Mol. Cell. Biol. 1993, 13. [Google Scholar] [CrossRef]
- Li, Z.; Bailey, A.D.; Buchowski, J.; Weiner, A.M. A Tandem Array of Minimal U1 Small Nuclear RNA Genes is Sufficient to Generate a New Adenovirus Type 12Inducible Chromosome Fragile Site. J. Virol. 1998, 72, 4205–4211. [Google Scholar] [CrossRef] [PubMed]
- Caporossi, D.; Bacchetti, S.; Kell, B.; Jewers, R.J.; Cason, J.; Pakarian, F.; Kaye, J.N.; Best, J.M. Definition of adenovirus type 5 functions involved in the induction of chromosomal aberrations in human cells. J. Gen. Virol. 1990, 71, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Caporossi, D.; Bacchetti, S.; Nicoletti, B. Synergism between aphidicolin and adenoviruses in the induction of breaks at fragile sites on human chromosomes. Cancer Genet. Cytogenet. 1991, 54, 39–53. [Google Scholar] [CrossRef]
- Durnam, D.M.; Smith, P.P.; Menninger, J.C.; McDougall, J.K. The El region of human adenovirus type 12 determines the sites of virally induced chromosomal damage. In Cancer Cells 4: DNA Tumor Viruses; Botchan, M., Ed.; Cold Spring Harbor Laboratory: Huntington, NY, USA, 1986; pp. 349–354. [Google Scholar]
- Schramayr, S.; Caporossi, D.; Mak, I.; Jelinek, T.; Bacchetti, S. Chromosomal damage induced by human adenovirus type 12 requires expression of the E1B 55-kilodalton viral protein. J. Virol. 1990, 64, 2090–2095. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Grand, R.J.A. Adenovirus E1B 55-Kilodalton Protein: Multiple Roles in Viral Infection and Cell Transformation. J. Virol. 2009, 83, 4000–4012. [Google Scholar] [CrossRef]
- Schreiner, S.; Wimmer, P.; Dobner, T. Adenovirus degradation of cellular proteins. Futur. Microbiol. 2012, 7, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, P.; Ip, W.H.; Dobner, T.; Gonzalez, R.A. The biology of the adenovirus E1B 55K protein. FEBS Lett. 2019, 593, 3504–3517. [Google Scholar] [CrossRef]
- Kleinberger, T. En Guard! The Interactions between Adenoviruses and the DNA Damage Response. Viruses 2020, 12, 996. [Google Scholar] [CrossRef]
- Herrmann, C.; Dybas, J.M.; Liddle, J.C.; Price, A.M.; Hayer, K.E.; Lauman, R.; Purman, C.E.; Charman, M.; Kim, E.T.; Garcia, B.A.; et al. Adenovirus-mediated ubiquitination alters protein–RNA binding and aids viral RNA processing. Nat. Microbiol. 2020, 5, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Orazio, N.I.; Naeger, C.M.; Karlseder, J.; Weitzman, M.D. The Adenovirus E1b55K/E4orf6 Complex Induces Degradation of the Bloom Helicase during Infection. J. Virol. 2010, 85, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Rohleder, K.J.; Hanakahi, L.A.; Ketner, G. Adenovirus E4 34k and E1b 55k Oncoproteins Target Host DNA Ligase IV for Proteasomal Degradation. J. Virol. 2007, 81, 7034–7040. [Google Scholar] [CrossRef]
- Stracker, T.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11–Rad50–NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef]
- Sarnow, P.; Ho, Y.S.; Williams, J.; Levine, A.J. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 1982, 28, 387–394. [Google Scholar] [CrossRef]
- Hagkarim, N.C.; Ryan, E.L.; Byrd, P.J.; Hollingworth, R.; Shimwell, N.J.; Agathanggelou, A.; Vavasseur, M.; Kolbe, V.; Speiseder, T.; Dobner, T.; et al. Degradation of a Novel DNA Damage Response Protein, Tankyrase 1 Binding Protein 1, following Adenovirus Infection. J. Virol. 2018, 92, e02034-17. [Google Scholar] [CrossRef]
- Gupta, A.; Jha, S.; Engel, D.A.; Ornelles, D.A.; Dutta, A. Tip60 degradation by adenovirus relieves transcriptional repression of viral transcriptional activator EIA. Oncogene 2012, 32, 5017–5025. [Google Scholar] [CrossRef]
- Blackford, A.N.; Patel, R.N.; Forrester, N.A.; Theil, K.; Groitl, P.; Stewart, G.; Taylor, A.M.R.; Morgan, I.M.; Dobner, T.; Grand, R.J.A.; et al. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc. Natl. Acad. Sci. USA 2010, 107, 12251–12256. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.T.; Schwartz, R.A.; Stracker, T.; Lilley, C.E.; Lee, D.V.; Weitzman, M.D. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003, 22, 6610–6620. [Google Scholar] [CrossRef] [PubMed]
- Karen, K.A.; Hoey, P.J.; Young, C.S.H.; Hearing, P. Temporal Regulation of the Mre11-Rad50-Nbs1 Complex during Adenovirus Infection. J. Virol. 2009, 83, 4565–4573. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA Damage and Repair Pathways by Human Tumour Viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Charman, M.; Weitzman, M.D.; Charman, M.; Weitzman, M.D. Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location. Viruses 2020, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, P.H.; Lecane, P.S.; Roberts, S.; Rookes, S.M.; Grand, R.J.; Parkhill, J. Adenovirus type 12 early region 1B 54K protein significantly extends the life span of normal mammalian cells in culture. J. Virol. 1997, 71, 6629–6640. [Google Scholar] [CrossRef]
- Byrd, P.; Brown, K.W.; Gallimore, P.H. Malignant transformation of human embryo retinoblasts by cloned adenovirus 12 DNA. Nature 1982, 298, 69–71. [Google Scholar] [CrossRef]
- Mendez, J.; Stillman, B. Chromatin Association of Human Origin Recognition Complex, Cdc6, and Minichromosome Maintenance Proteins during the Cell Cycle: Assembly of Prereplication Complexes in Late Mitosis. Mol. Cell. Biol. 2000, 20, 8602–8612. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef]
- Henry-Mowatt, J.; Jackson, D.; Masson, J.Y.; Johnson, P.A.; Clements, P.M.; Benson, F.E.; Thompson, L.H.; Takeda, S.; West, S.C.; Caldecott, K.W. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol. Cell 2003, 11, 1109–1117. [Google Scholar] [CrossRef]
- Nieminuszczy, J.; Schwab, R.A.; Niedzwiedz, W. The DNA fibre technique–tracking helicases at work. Methods 2016, 108, 92–98. [Google Scholar] [CrossRef]
- Boguslawski, S.J.; Smith, D.E.; Michalak, M.A.; Mickelson, K.E.; Yehle, C.O.; Patterson, W.L.; Carrico, R.J. Characterization of monoclonal antibody to DNA RNA and its application to immunodetection of hybrids. J. Immunol. Methods 1986, 89, 123–130. [Google Scholar] [CrossRef]
- Forrester, N.A.; Sedgwick, G.G.; Thomas, A.; Blackford, A.; Speiseder, T.; Dobner, T.; Byrd, P.J.; Stewart, G.S.; Turnell, A.S.; Grand, R.J.A. Serotype-Specific Inactivation of the Cellular DNA Damage Response during Adenovirus Infection. J. Virol. 2010, 85, 2201–2211. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Gilson, T.; Dallaire, F.; Ketner, G.; Branton, P.E.; Blanchette, P. The E4orf6/E1B55K E3 Ubiquitin Ligase Complexes of Human Adenoviruses Exhibit Heterogeneity in Composition and Substrate Specificity. J. Virol. 2010, 85, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Grand, R.J.; Owen, D.; Rookes, S.M.; Gallimore, P.H. Control of p53 Expression by Adenovirus 12 Early Region 1A and Early Region 1B 54K Proteins. Virology 1996, 218, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Gargano, S.; Wang, P.; Rusanganwa, E.; Bacchetti, S. The transcriptionally competent U2 gene is necessary and sufficient for adenovirus type 12 induction of the fragile site at 17q21-22. Mol. Cell. Biol. 1995, 15, 6256–6261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liao, D.; Yu, A.; Weiner, A.M. Coexpression of the Adenovirus 12 E1B 55 kDa Oncoprotein and Cellular Tumor Suppressor p53 Is Sufficient to Induce Metaphase Fragility of the HumanRNU2Locus. Virology 1999, 254, 11–23. [Google Scholar] [CrossRef][Green Version]
- Whittaker, J.L.; Byrd, P.J.; Grand, R.J.; Gallimore, P.H. Isolation and characterization of four adenovirus type 12-transformed human embryo kidney cell lines. Mol. Cell. Biol. 1984, 4. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Said, M.; Naim, V. DNA Replication Stress and Chromosomal Instability: Dangerous Liaisons. Genes 2020, 11, 642. [Google Scholar] [CrossRef]
- Schwartz, R.A.; Lakdawala, S.S.; Eshleman, H.D.; Russell, M.R.; Carson, C.T.; Weitzman, M.D. Distinct Requirements of Adenovirus E1b55K Protein for Degradation of Cellular Substrates. J. Virol. 2008, 82, 9043–9055. [Google Scholar] [CrossRef]
- Yew, P.R.; Berk, A.J. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 1992, 357, 82–85. [Google Scholar] [CrossRef]
- Schreiner, S.; Wimmer, P.; Sirma, H.; Everett, R.D.; Blanchette, P.; Groitl, P.; Dobner, T. Proteasome-Dependent Degradation of Daxx by the Viral E1B-55K Protein in Human Adenovirus-Infected Cells. J. Virol. 2010, 84, 7029–7038. [Google Scholar] [CrossRef]
- Zantema, A.; Fransen, J.; Davis-Olivier, A.; Ramaekers, F.C.; Vooijs, G.P.; Deleys, B.; Van Der Eb, A.J. Localization of the E1 B proteins of adenovirus 5 in transformed cells, as revealed by interaction with monoclonal antibodies. Virology 1985, 142, 44–58. [Google Scholar] [CrossRef]
- Araujo, F.D.; Stracker, T.H.; Carson, C.T.; Lee, D.V.; Weitzman, M.D. Adenovirus type 5 e4orf3 protein targets the mre11 complex to cytoplasmic aggresomes. J. Virol. 2005, 79, 11382–11391. [Google Scholar] [CrossRef]
- Liu, Y.; Shevchenko, A.; Shevchenko, A.; Berk, A.J. Adenovirus Exploits the Cellular Aggresome Response to Accelerate Inactivation of the MRN Complex. J. Virol. 2005, 79, 14004–14016. [Google Scholar] [CrossRef]
- Löber, C.; Lenz-Stöppler, C.; Dobbelstein, M. Adenovirus E1-transformed cells grow despite the continuous presence of transcriptionally active p53. J. Gen. Virol. 2002, 83, 2047–2057. [Google Scholar] [CrossRef][Green Version]
- Sieber, T.; Dobner, T. Adenovirus Type 5 Early Region 1B 156R Protein Promotes Cell Transformation Independently of Repression of p53-Stimulated Transcription. J. Virol. 2007, 81, 95–105. [Google Scholar] [CrossRef]
- Härtl, B.; Zeller, T.; Blanchette, P.; Kremmer, E.; Dobner, T. Adenovirus type 5 early region 1B 55-kDa oncoprotein can promote cell transformation by a mechanism independent from blocking p53-activated transcription. Oncogene 2008, 27, 3673–3684. [Google Scholar] [CrossRef]
- Ip, W.H.; Dobner, T. Cell transformation by the adenovirus oncogenes E1 and E4. FEBS Lett. 2020, 594, 1848–1860. [Google Scholar] [CrossRef]
- Houweling, A.; van den Elsen, P.J.; Van Der Eb, A.J. Partial transformation of primary rat cells by the leftmost 4.5% fragment of adenovirus 5 DNA. Virology 1980, 105, 537–550. [Google Scholar] [CrossRef]
- Van den Elsen, P.; Houweling, A.; Van Der Eb, A. Expression of region E1b of human adenoviruses in the absence of region E1a is not sufficient for complete transformation. Virology 1983, 128, 377–390. [Google Scholar] [CrossRef]
- Gallimore, P.H.; Byrd, P.J.; Whittaker, J.L.; Grand, R.J. Properties of rat cells transformed by DNA plasmids containing adenovirus type 12 E1 DNA or specific fragments of the E1 region: Comparison of transforming frequencies. Cancer Res. 1985, 45, 2670–2680. [Google Scholar]
- Bernards, R.; Houweling, A.; Schrier, P.I.; Bos, J.L.; Van Der Eb, A.J. Characterization of cells transformed by Ad5/Ad12 hybrid early region I plasmids. Virology 1982, 120, 422–432. [Google Scholar] [CrossRef]
- Bernards, R.; Schrier, P.I.; Bos, J.L.; Van Der Eb, A.J. Role of adenovirus types 5 and 12 early region 1 b tumor antigens in oncogenic transformation. Virology 1983, 127, 45–53. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abualfaraj, T.; Hagkarim, N.C.; Hollingworth, R.; Grange, L.; Jhujh, S.; Stewart, G.S.; Grand, R.J. The Promotion of Genomic Instability in Human Fibroblasts by Adenovirus 12 Early Region 1B 55K Protein in the Absence of Viral Infection. Viruses 2021, 13, 2444. https://doi.org/10.3390/v13122444
Abualfaraj T, Hagkarim NC, Hollingworth R, Grange L, Jhujh S, Stewart GS, Grand RJ. The Promotion of Genomic Instability in Human Fibroblasts by Adenovirus 12 Early Region 1B 55K Protein in the Absence of Viral Infection. Viruses. 2021; 13(12):2444. https://doi.org/10.3390/v13122444
Chicago/Turabian StyleAbualfaraj, Tareq, Nafiseh Chalabi Hagkarim, Robert Hollingworth, Laura Grange, Satpal Jhujh, Grant S. Stewart, and Roger J. Grand. 2021. "The Promotion of Genomic Instability in Human Fibroblasts by Adenovirus 12 Early Region 1B 55K Protein in the Absence of Viral Infection" Viruses 13, no. 12: 2444. https://doi.org/10.3390/v13122444
APA StyleAbualfaraj, T., Hagkarim, N. C., Hollingworth, R., Grange, L., Jhujh, S., Stewart, G. S., & Grand, R. J. (2021). The Promotion of Genomic Instability in Human Fibroblasts by Adenovirus 12 Early Region 1B 55K Protein in the Absence of Viral Infection. Viruses, 13(12), 2444. https://doi.org/10.3390/v13122444