
Metabolic Modifications by Common Respiratory Viruses and Their Potential as New Antiviral Targets

 , and
, and 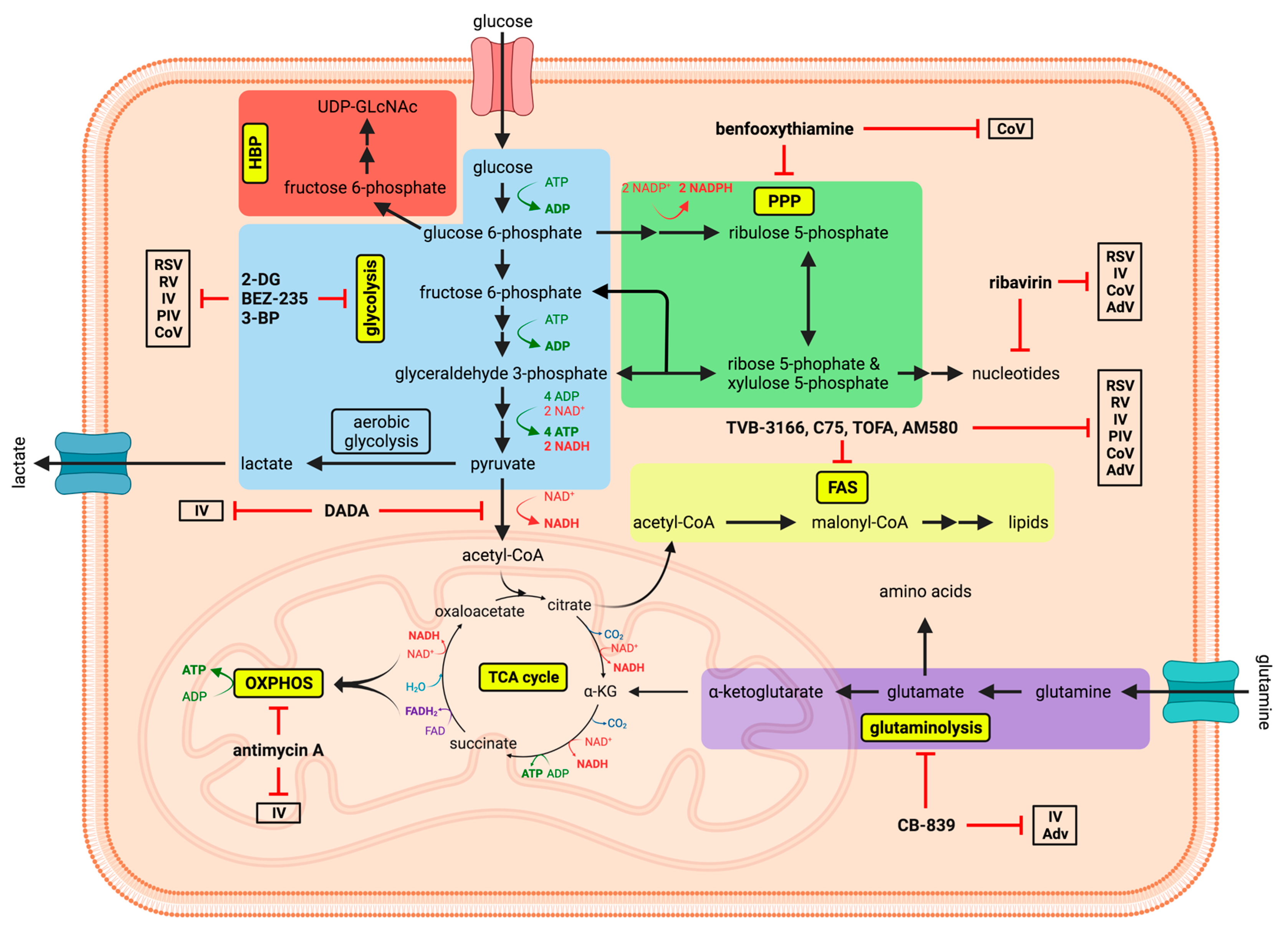{kind=link}
Abstract
1. Introduction
2. Respiratory Syncytial Viruses
3. Rhinoviruses
4. Influenza Viruses
5. Parainfluenza Viruses
6. Coronaviruses
7. Adenoviruses
8. Therapeutic Potential of Metabolic Interference—What It Is and What It Might Become?
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galeas-Pena, M.; McLaughlin, N.; Pociask, D. The role of the innate immune system on pulmonary infections. Biol. Chem. 2019, 400, 443–456. [Google Scholar] [CrossRef]
- Shi, T.; Arnott, A.; Semogas, I.; Falsey, A.R.; Openshaw, P.; Wedzicha, J.A.; Campbell, H.; Nair, H.; Investigators, R. The Etiological Role of Common Respiratory Viruses in Acute Respiratory Infections in Older Adults: A Systematic Review and Meta-analysis. J. Infect. Dis. 2020, 222, S563–S569. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Christiaansen, A.; Varga, S.M.; Spencer, J.V. Viral manipulation of the host immune response. Curr. Opin. Immunol. 2015, 36, 54–60. [Google Scholar] [CrossRef]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, 369, 706–712. [Google Scholar] [CrossRef]
- Ivanisenko, N.V.; Seyrek, K.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. The role of death domain proteins in host response upon SARS-CoV-2 infection: Modulation of programmed cell death and translational applications. Cell Death Discov. 2020, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.J. Viruses and apoptosis. Int. J. Exp. Pathol. 2001, 82, 65–76. [Google Scholar] [CrossRef]
- Preugschas, H.F.; Hrincius, E.R.; Mewis, C.; Tran, G.V.Q.; Ludwig, S.; Ehrhardt, C. Late activation of the Raf/MEK/ERK pathway is required for translocation of the respiratory syncytial virus F protein to the plasma membrane and efficient viral replication. Cell Microbiol. 2019, 21, e12955. [Google Scholar] [CrossRef]
- Schreiber, A.; Boff, L.; Anhlan, D.; Krischuns, T.; Brunotte, L.; Schuberth, C.; Wedlich-Soldner, R.; Drexler, H.; Ludwig, S. Dissecting the mechanism of signaling-triggered nuclear export of newly synthesized influenza virus ribonucleoprotein complexes. Proc. Natl. Acad. Sci. USA 2020, 117, 16557–16566. [Google Scholar] [CrossRef] [PubMed]
- Grimes, J.M.; Grimes, K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Borgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J. Biol. Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, J.; Bartlett, E.; Schomacker, H.; Surman, S.; Akira, S.; Bae, Y.S.; Collins, P.; Murphy, B.; Schmidt, A. The C proteins of human parainfluenza virus type 1 limit double-stranded RNA accumulation that would otherwise trigger activation of MDA5 and protein kinase R. J. Virol. 2011, 85, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Berberich-Siebelt, F.; Wolff, T.; Walczak, H.; Planz, O.; Ludwig, S. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J. Biol. Chem. 2004, 279, 30931–30937. [Google Scholar] [CrossRef]
- Mazur, I.; Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Puthavathana, P.; Silberzahn, T.; Wolff, T.; Planz, O.; Ludwig, S. Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-kappaB-inhibiting activity. Cell Microbiol. 2007, 9, 1683–1694. [Google Scholar] [CrossRef]
- Droebner, K.; Haasbach, E.; Dudek, S.E.; Scheuch, G.; Nocker, K.; Canisius, S.; Ehrhardt, C.; von Degenfeld, G.; Ludwig, S.; Planz, O. Pharmacodynamics, Pharmacokinetics, and Antiviral Activity of BAY 81-8781, a Novel NF-kappaB Inhibiting Anti-influenza Drug. Front. Microbiol. 2017, 8, 2130. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Kwon, H.; Genin, P. Hostile takeovers: Viral appropriation of the NF-kappaB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef]
- Gualdoni, G.A.; Mayer, K.A.; Kapsch, A.M.; Kreuzberg, K.; Puck, A.; Kienzl, P.; Oberndorfer, F.; Fruhwirth, K.; Winkler, S.; Blaas, D.; et al. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc. Natl. Acad. Sci. USA 2018, 115, E7158–E7165. [Google Scholar] [CrossRef]
- Beziau, A.; Brand, D.; Piver, E. The Role of Phosphatidylinositol Phosphate Kinases during Viral Infection. Viruses 2020, 12, 1124. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Ludwig, S. A new player in a deadly game: Influenza viruses and the PI3K/Akt signalling pathway. Cell Microbiol. 2009, 11, 863–871. [Google Scholar] [CrossRef]
- Morris, D.R.; Qu, Y.; Agrawal, A.; Garofalo, R.P.; Casola, A. HIF-1alpha Modulates Core Metabolism and Virus Replication in Primary Airway Epithelial Cells Infected with Respiratory Syncytial Virus. Viruses 2020, 12, 1088. [Google Scholar] [CrossRef]
- Reyes, A.; Corrales, N.; Galvez, N.M.S.; Bueno, S.M.; Kalergis, A.M.; Gonzalez, P.A. Contribution of hypoxia inducible factor-1 during viral infections. Virulence 2020, 11, 1482–1500. [Google Scholar] [CrossRef]
- Smallwood, H.S.; Duan, S.; Morfouace, M.; Rezinciuc, S.; Shulkin, B.L.; Shelat, A.; Zink, E.E.; Milasta, S.; Bajracharya, R.; Oluwaseum, A.J.; et al. Targeting Metabolic Reprogramming by Influenza Infection for Therapeutic Intervention. Cell Rep. 2017, 19, 1640–1653. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Liang, X.; Hou, J.; Aisa, Y.; Wu, H.; Zhang, Z.; Nuermaimaiti, N.; Zhao, Y.; Jiang, S.; Guan, Y. Adenovirus type 36 regulates adipose stem cell differentiation and glucolipid metabolism through the PI3K/Akt/FoxO1/PPARgamma signaling pathway. Lipids Health Dis. 2019, 18, 70. [Google Scholar] [CrossRef]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Ruuskanen, O.; Lahti, E.; Jennings, L.C.; Murdoch, D.R. Viral pneumonia. Lancet 2011, 377, 1264–1275. [Google Scholar] [CrossRef]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef]
- Walsh, E.E. Respiratory Syncytial Virus Infection: An Illness for All Ages. Clin. Chest Med. 2017, 38, 29–36. [Google Scholar] [CrossRef]
- Varkey, J.B.; Varkey, B. Viral infections in patients with chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med. 2008, 14, 89–94. [Google Scholar] [CrossRef]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Feldman, A.S.; He, Y.; Moore, M.L.; Hershenson, M.B.; Hartert, T.V. Toward primary prevention of asthma. Reviewing the evidence for early-life respiratory viral infections as modifiable risk factors to prevent childhood asthma. Am. J. Respir. Crit. Care Med. 2015, 191, 34–44. [Google Scholar] [CrossRef] [PubMed]
- The IMpact-RSV Study Group. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 1998, 102, 531–537. [Google Scholar] [CrossRef]
- Martin-Vicente, M.; Gonzalez-Riano, C.; Barbas, C.; Jimenez-Sousa, M.A.; Brochado-Kith, O.; Resino, S.; Martinez, I. Metabolic changes during respiratory syncytial virus infection of epithelial cells. PLoS ONE 2020, 15, e0230844. [Google Scholar] [CrossRef] [PubMed]
- DeVincenzo, J.; Lambkin-Williams, R.; Wilkinson, T.; Cehelsky, J.; Nochur, S.; Walsh, E.; Meyers, R.; Gollob, J.; Vaishnaw, A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2010, 107, 8800–8805. [Google Scholar] [CrossRef]
- Martinez, I.; Lombardia, L.; Garcia-Barreno, B.; Dominguez, O.; Melero, J.A. Distinct gene subsets are induced at different time points after human respiratory syncytial virus infection of A549 cells. J. Gen. Virol. 2007, 88, 570–581. [Google Scholar] [CrossRef]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Alsuwaidi, A.R.; Benedict, S.; Kochiyil, J.; Mustafa, F.; Hartwig, S.M.; Almarzooqi, S.; Albawardi, A.; Rizvi, T.A.; Varga, S.M.; Souid, A.K. Bioenergetics of murine lungs infected with respiratory syncytial virus. Virol. J. 2013, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, T.J.; Hodes, D.S.; Gerin, J.; Camargo, E.; Chanock, R.M. Effect of 2-deoxy-D-glucose and glucosamine on the growth and functions of respiratory syncytial and parainfluenza 3 viruses. Virology 1975, 67, 306–309. [Google Scholar] [CrossRef]
- Turi, K.N.; Romick-Rosendale, L.; Gebretsadik, T.; Watanabe, M.; Brunwasser, S.; Anderson, L.J.; Moore, M.L.; Larkin, E.K.; Peebles, R.S., Jr.; Hartert, T.V. Using urine metabolomics to understand the pathogenesis of infant respiratory syncytial virus (RSV) infection and its role in childhood wheezing. Metabolomics 2018, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Adamko, D.J.; Saude, E.; Bear, M.; Regush, S.; Robinson, J.L. Urine metabolomic profiling of children with respiratory tract infections in the emergency department: A pilot study. BMC Infect. Dis. 2016, 16, 439. [Google Scholar] [CrossRef]
- Rezinciuc, S.; Bezavada, L.; Bahadoran, A.; Ingels, J.F.; Kim, Y.-Y.; Cormier, S.A.; Shulkin, B.L.; Devincenzo, J.P.; Smallwood, H.S. Respiratory syncytial virus induces hypermetabolism in pediatric airways. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar]
- Martinez, I.; Garcia-Carpizo, V.; Guijarro, T.; Garcia-Gomez, A.; Navarro, D.; Aranda, A.; Zambrano, A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016, 7, 427–442. [Google Scholar] [CrossRef]
- Hu, M.; Schulze, K.E.; Ghildyal, R.; Henstridge, D.C.; Kolanowski, J.L.; New, E.J.; Hong, Y.; Hsu, A.C.; Hansbro, P.M.; Wark, P.A.; et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. eLife 2019, 8, e42448. [Google Scholar] [CrossRef]
- Ohol, Y.M.; Wang, Z.; Kemble, G.; Duke, G. Direct Inhibition of Cellular Fatty Acid Synthase Impairs Replication of Respiratory Syncytial Virus and Other Respiratory Viruses. PLoS ONE 2015, 10, e0144648. [Google Scholar] [CrossRef]
- Waman, V.P.; Kolekar, P.S.; Kale, M.M.; Kulkarni-Kale, U. Population structure and evolution of Rhinoviruses. PLoS ONE 2014, 9, e88981. [Google Scholar] [CrossRef]
- Calvo, C.; Casas, I.; Garcia-Garcia, M.L.; Pozo, F.; Reyes, N.; Cruz, N.; Garcia-Cuenllas, L.; Perez-Brena, P. Role of rhinovirus C respiratory infections in sick and healthy children in Spain. Pediatr. Infect. Dis. J. 2010, 29, 717–720. [Google Scholar] [CrossRef]
- Seemungal, T.; Harper-Owen, R.; Bhowmik, A.; Moric, I.; Sanderson, G.; Message, S.; Maccallum, P.; Meade, T.W.; Jeffries, D.J.; Johnston, S.L.; et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 1618–1623. [Google Scholar] [CrossRef]
- Nguyen, A.; Guedan, A.; Mousnier, A.; Swieboda, D.; Zhang, Q.; Horkai, D.; Le Novere, N.; Solari, R.; Wakelam, M.J.O. Host lipidome analysis during rhinovirus replication in HBECs identifies potential therapeutic targets. J. Lipid Res. 2018, 59, 1671–1684. [Google Scholar] [CrossRef]
- Roulin, P.S.; Lotzerich, M.; Torta, F.; Tanner, L.B.; van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef]
- Grassme, H.; Riehle, A.; Wilker, B.; Gulbins, E. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J. Biol. Chem. 2005, 280, 26256–26262. [Google Scholar] [CrossRef]
- Dreschers, S.; Franz, P.; Dumitru, C.; Wilker, B.; Jahnke, K.; Gulbins, E. Infections with human rhinovirus induce the formation of distinct functional membrane domains. Cell Physiol. Biochem. 2007, 20, 241–254. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. The pathology of influenza virus infections. Annu. Rev. Pathol. 2008, 3, 499–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, J.; Huang, K.; Li, K.S.; Yuen, K.Y.; Guan, Y.; Chen, H.; Ng, W.F. Systemic infection of avian influenza A virus H5N1 subtype in humans. Hum. Pathol. 2009, 40, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Speers, D.J.; Moss, D.M.; Minney-Smith, C.; Levy, A.; Smith, D.W. Influenza and respiratory syncytial virus are the major respiratory viruses detected from prospective testing of pediatric and adult coronial autopsies. Influenza Other Respir. Viruses 2013, 7, 1113–1121. [Google Scholar] [CrossRef]
- Principi, N.; Camilloni, B.; Alunno, A.; Polinori, I.; Argentiero, A.; Esposito, S. Drugs for Influenza Treatment: Is There Significant News? Front. Med. 2019, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Ainai, A.; Suzuki, T.; Hasegawa, H. The road to a more effective influenza vaccine: Up to date studies and future prospects. Vaccine 2017, 35, 5388–5395. [Google Scholar] [CrossRef]
- Daniels, J.B.; Eaton, M.D.; Perry, M.E. Effect of glucose on the growth of influenza virus in deembryonated eggs and tissue cultures. J. Immunol. 1952, 69, 321–329. [Google Scholar] [PubMed]
- Klemperer, H. Glucose breakdown in chick embryo cells infected with influenza virus. Virology 1961, 13, 68–77. [Google Scholar] [CrossRef]
- Ritter, J.B.; Wahl, A.S.; Freund, S.; Genzel, Y.; Reichl, U. Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling. BMC Syst. Biol. 2010, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Kuss-Duerkop, S.K.; Wang, J.; Mena, I.; White, K.; Metreveli, G.; Sakthivel, R.; Mata, M.A.; Munoz-Moreno, R.; Chen, X.; Krammer, F.; et al. Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late stage replication. PLoS Pathog. 2017, 13, e1006635. [Google Scholar] [CrossRef] [PubMed]
- Hrincius, E.R.; Dierkes, R.; Anhlan, D.; Wixler, V.; Ludwig, S.; Ehrhardt, C. Phosphatidylinositol-3-kinase (PI3K) is activated by influenza virus vRNA via the pathogen pattern receptor Rig-I to promote efficient type I interferon production. Cell Microbiol. 2011, 13, 1907–1919. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 2007, 88, 13–18. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, K.; Min, J.; Chen, C.; Cao, Y.; Ding, C.; Liu, W.; Li, J. Metabolomic Analysis of Influenza A Virus A/WSN/1933 (H1N1) Infected A549 Cells during First Cycle of Viral Replication. Viruses 2019, 11, 1007. [Google Scholar] [CrossRef] [PubMed]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Ting Wu, T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef] [PubMed]
- Kohio, H.P.; Adamson, A.L. Glycolytic control of vacuolar-type ATPase activity: A mechanism to regulate influenza viral infection. Virology 2013, 444, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Kilbourne, E.D. Inhibition of influenza virus multiplication with a glucose antimetabolite (2-deoxy-D-glucose). Nature 1959, 183, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Deinhardt-Emmer, S.; Jackel, L.; Haring, C.; Bottcher, S.; Wilden, J.J.; Gluck, B.; Heller, R.; Schmidtke, M.; Koch, M.; Loffler, B.; et al. Inhibition of Phosphatidylinositol 3-Kinase by Pictilisib Blocks Influenza Virus Propagation in Cells and in Lungs of Infected Mice. Biomolecules 2021, 11, 808. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Indalao, I.L.; Chida, J.; Yamamoto, Y.; Hanawa, M.; Kido, H. Diisopropylamine dichloroacetate, a novel pyruvate dehydrogenase kinase 4 inhibitor, as a potential therapeutic agent for metabolic disorders and multiorgan failure in severe influenza. PLoS ONE 2014, 9, e98032. [Google Scholar] [CrossRef]
- Nakamura, K.; Compans, R.W. Effects of glucosamine, 2-deoxyglucose, and tunicamycin on glycosylation, sulfation, and assembly of influenza viral proteins. Virology 1978, 84, 303–319. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Heaton, N.S.; Randall, G. Lipids at the interface of virus-host interactions. Curr. Opin. Microbiol. 2012, 15, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Liu, N.; Yang, Z.; Song, W.; Wang, P.; Chen, H.; Lucio, M.; Schmitt-Kopplin, P.; Chen, G.; Cai, Z. GC/MS-based metabolomics reveals fatty acid biosynthesis and cholesterol metabolism in cell lines infected with influenza A virus. Talanta 2010, 83, 262–268. [Google Scholar] [CrossRef]
- Tisoncik-Go, J.; Gasper, D.J.; Kyle, J.E.; Eisfeld, A.J.; Selinger, C.; Hatta, M.; Morrison, J.; Korth, M.J.; Zink, E.M.; Kim, Y.M.; et al. Integrated Omics Analysis of Pathogenic Host Responses during Pandemic H1N1 Influenza Virus Infection: The Crucial Role of Lipid Metabolism. Cell Host Microbe 2016, 19, 254–266. [Google Scholar] [CrossRef]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef]
- Limsuwat, N.; Boonarkart, C.; Phakaratsakul, S.; Suptawiwat, O.; Auewarakul, P. Influence of cellular lipid content on influenza A virus replication. Arch. Virol. 2020, 165, 1151–1161. [Google Scholar] [CrossRef]
- Yuan, S.; Chu, H.; Chan, J.F.; Ye, Z.W.; Wen, L.; Yan, B.; Lai, P.M.; Tee, K.M.; Huang, J.; Chen, D.; et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019, 10, 120. [Google Scholar] [CrossRef]
- Ivanova, P.T.; Myers, D.S.; Milne, S.B.; McClaren, J.L.; Thomas, P.G.; Brown, H.A. Lipid composition of viral envelope of three strains of influenza virus—Not all viruses are created equal. ACS Infect. Dis. 2015, 1, 399–452. [Google Scholar] [CrossRef] [PubMed]
- Demers, A.; Ran, Z.; Deng, Q.; Wang, D.; Edman, B.; Lu, W.; Li, F. Palmitoylation is required for intracellular trafficking of influenza B virus NB protein and efficient influenza B virus growth in vitro. J. Gen. Virol. 2014, 95, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, H.; Bai, L.; Yu, Y.; Sun, Z.; Yan, Y.; Zhou, J. Mitochondrial proteomic analysis of human host cells infected with H3N2 swine influenza virus. J. Proteom. 2013, 91, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, W.W.; Kleinschmidt, E. Concerning the relation of the Krebs cycle to virus propagation. J. Biol. Chem. 1951, 189, 421–428. [Google Scholar] [CrossRef]
- Bahadoran, A.; Bezavada, L.; Smallwood, H.S. Fueling influenza and the immune response: Implications for metabolic reprogramming during influenza infection and immunometabolism. Immunol. Rev. 2020, 295, 140–166. [Google Scholar] [CrossRef]
- Vaughan, R.A.; Garcia-Smith, R.; Trujillo, K.A.; Bisoffi, M. Tumor necrosis factor alpha increases aerobic glycolysis and reduces oxidative metabolism in prostate epithelial cells. Prostate 2013, 73, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.D.; Hu, X.; Ko, E.J.; Park, S.; Lee, Y.T.; Orr, M.; Fernandes, J.; Uppal, K.; Kang, S.M.; Jones, D.P.; et al. Metabolic pathways of lung inflammation revealed by high-resolution metabolomics (HRM) of H1N1 influenza virus infection in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R906–R916. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Rathmell, J.C. Metabolic Signaling Drives IFN-gamma. Cell Metab. 2016, 24, 651–652. [Google Scholar] [CrossRef][Green Version]
- Fritsch, S.D.; Weichhart, T. Effects of Interferons and Viruses on Metabolism. Front. Immunol. 2016, 7, 630. [Google Scholar] [CrossRef]
- Branche, A.R.; Falsey, A.R. Parainfluenza Virus Infection. Semin. Respir. Crit. Care Med. 2016, 37, 538–554. [Google Scholar] [CrossRef]
- Schmidt, A.C.; Schaap-Nutt, A.; Bartlett, E.J.; Schomacker, H.; Boonyaratanakornkit, J.; Karron, R.A.; Collins, P.L. Progress in the development of human parainfluenza virus vaccines. Expert Rev. Respir. Med. 2011, 5, 515–526. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kato, J.; Kohara, M.; Galinski, M.S. Antiviral effects of glycosylation and glucose trimming inhibitors on human parainfluenza virus type 3. Antivir. Res. 2006, 72, 1–9. [Google Scholar] [CrossRef]
- Klenk, H.D.; Choppin, P.W. Plasma membrane lipids and parainfluenza virus assembly. Virology 1970, 40, 939–947. [Google Scholar] [CrossRef]
- Rabbani, M.A.G.; Barik, S. 5-Hydroxytryptophan, a major product of tryptophan degradation, is essential for optimal replication of human parainfluenza virus. Virology 2017, 503, 46–51. [Google Scholar] [CrossRef]
- Li, Y.D.; Chi, W.Y.; Su, J.H.; Ferrall, L.; Hung, C.F.; Wu, T.C. Coronavirus vaccine development: From SARS and MERS to COVID-19. J. Biomed. Sci. 2020, 27, 104. [Google Scholar] [CrossRef]
- van der Hoek, L. Human coronaviruses: What do they cause? Antivir Ther. 2007, 12, 651–658. [Google Scholar]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef]
- Del Rio, C.; Collins, L.F.; Malani, P. Long-term Health Consequences of COVID-19. JAMA 2020, 324, 1723–1724. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.; Chan, J.F.; et al. Characterization of the Lipidomic Profile of Human Coronavirus-Infected Cells: Implications for Lipid Metabolism Remodeling upon Coronavirus Replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Silvas, J.A.; Jureka, A.S.; Nicolini, A.M.; Chvatal, S.A.; Basler, C.F. Inhibitors of VPS34 and lipid metabolism suppress SARS-CoV-2 replication. bioRxiv 2020. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Hardt, M.; Schwudke, D.; Neuman, B.W.; Pleschka, S.; Ziebuhr, J. Inhibition of Cytosolic Phospholipase A2alpha Impairs an Early Step of Coronavirus Replication in Cell Culture. J. Virol. 2018, 92, e01463-17. [Google Scholar] [CrossRef]
- Nagy, P.D.; Strating, J.R.; van Kuppeveld, F.J. Building Viral Replication Organelles: Close Encounters of the Membrane Types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Munch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.N.; Kumar, A.; Rai, Y.; Kumari, N.; Vedagiri, D.; Harshan, K.H.; Chinnadurai, V.; Chandna, S. Glycolytic inhibitor 2-Deoxy-D-glucose attenuates SARS-CoV-2 multiplication in host cells and weakens the infective potential of progeny virions. bioRxiv 2021. [Google Scholar] [CrossRef]
- Balkrishna, A.; Thakur, P.; Singh, S.; Dev, S.N.C.; Jain, V.; Varshney, A.; Sharma, R.K. Glucose antimetabolite 2-Deoxy-D-Glucose and its derivative as promising candidates for tackling COVID-19: Insights derived from in silico docking and molecular simulations. Authorea 2021. [Google Scholar] [CrossRef]
- Bojkova, D.; Costa, R.; Bechtel, M.; Ciesek, S.; Michaelis, M.; Cinatl, J. Targeting pentose phosphate pathway for SARS-CoV-2 therapy. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; Denison, M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef]
- Cameron, C.E.; Castro, C. The mechanism of action of ribavirin: Lethal mutagenesis of RNA virus genomes mediated by the viral RNA-dependent RNA polymerase. Curr. Opin. Infect. Dis. 2001, 14, 757–764. [Google Scholar] [CrossRef]
- Ayres, J.S. A metabolic handbook for the COVID-19 pandemic. Nat. Metab. 2020, 2, 572–585. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Dalan, R.; Hopkins, D.; Mingrone, G.; Boehm, B.O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020, 16, 297–298. [Google Scholar] [CrossRef]
- Santos-Beneit, F.; Raskevicius, V.; Skeberdis, V.A.; Bordel, S. A metabolic modeling approach reveals promising therapeutic targets and antiviral drugs to combat COVID-19. Sci. Rep. 2021, 11, 11982. [Google Scholar] [CrossRef]
- Ismail, A.M.; Zhou, X.; Dyer, D.W.; Seto, D.; Rajaiya, J.; Chodosh, J. Genomic foundations of evolution and ocular pathogenesis in human adenovirus species D. FEBS Lett. 2019, 593, 3583–3608. [Google Scholar] [CrossRef]
- Georgi, F.; Kuttler, F.; Murer, L.; Andriasyan, V.; Witte, R.; Yakimovich, A.; Turcatti, G.; Greber, U.F. A high-content image-based drug screen of clinical compounds against cell transmission of adenovirus. Sci. Data 2020, 7, 265. [Google Scholar] [CrossRef]
- Scott, M.K.; Chommanard, C.; Lu, X.; Appelgate, D.; Grenz, L.; Schneider, E.; Gerber, S.I.; Erdman, D.D.; Thomas, A. Human Adenovirus Associated with Severe Respiratory Infection, Oregon, USA, 2013–2014. Emerg. Infect. Dis. 2016, 22, 1044–1051. [Google Scholar] [CrossRef]
- Lenaerts, L.; Naesens, L. Antiviral therapy for adenovirus infections. Antivir. Res. 2006, 71, 172–180. [Google Scholar] [CrossRef]
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef]
- Fisher, T.N.; Ginsberg, H.S. Accumulation of organic acids by HeLa cells infected with type 4 adenovirus. Proc. Soc. Exp. Biol. Med. 1957, 95, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Bardell, D. Glucose uptake and lactic acid production of adenovirus type 5-infected HEp-2 cells cultured under exponential growth and stationary phase conditions. Microbios 1977, 20, 139–144. [Google Scholar] [PubMed]
- Rozee, K.R.; Ottey, L.J.; Van Rooyen, C.E. Some metabolic effects of adenovirus infection in HeLa cells. Can. J. Microbiol. 1957, 3, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef]
- Silva, A.C.; Ana, P.T.; Paula, M.A. Impact of Adenovirus infection in host cell metabolism evaluated by (1)H-NMR spectroscopy. J. Biotechnol. 2016, 231, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Carinhas, N.; Koshkin, A.; Pais, D.A.; Alves, P.M.; Teixeira, A.P. (13) C-metabolic flux analysis of human adenovirus infection: Implications for viral vector production. Biotechnol. Bioeng. 2017, 114, 195–207. [Google Scholar] [CrossRef]
- Valdes, A.; Zhao, H.; Pettersson, U.; Lind, S.B. Time-resolved proteomics of adenovirus infected cells. PLoS ONE 2018, 13, e0204522. [Google Scholar] [CrossRef] [PubMed]
- Dyer, A.; Schoeps, B.; Frost, S.; Jakeman, P.; Scott, E.M.; Freedman, J.; Jacobus, E.J.; Seymour, L.W. Antagonism of Glycolysis and Reductive Carboxylation of Glutamine Potentiates Activity of Oncolytic Adenoviruses in Cancer Cells. Cancer Res. 2019, 79, 331–345. [Google Scholar] [CrossRef]
- McIntosh, K.; Payne, S.; Russell, W.C. Studies on lipid metabolism in cells infected with adenovirus. J. Gen. Virol. 1971, 10, 251–265. [Google Scholar] [CrossRef]
- Hidalgo, P.; Anzures, L.; Hernandez-Mendoza, A.; Guerrero, A.; Wood, C.D.; Valdes, M.; Dobner, T.; Gonzalez, R.A. Morphological, Biochemical, and Functional Study of Viral Replication Compartments Isolated from Adenovirus-Infected Cells. J. Virol. 2016, 90, 3411–3427. [Google Scholar] [CrossRef]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Lipidol. 2017, 28, 408–413. [Google Scholar] [CrossRef]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Siwiak, E.; Soltyka, M.; Priebe, A.; Zielinski, R.; Fokt, I.; Ziemniak, M.; Jaskiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, Z.; Zou, X.; Lan, Y.; Sun, X.; Wang, X.; Zhao, S.; Jiang, C.; Liu, H. Mechanisms underlying 3-bromopyruvate-induced cell death in colon cancer. J. Bioenerg. Biomembr. 2015, 47, 319–329. [Google Scholar] [CrossRef]
- Thomas, E.; Ghany, M.G.; Liang, T.J. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir. Chem. Chemother. 2012, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.S.; Creticos, C.M.; Jackson, G.G.; Bernstein, J.M.; Hayden, F.G.; Schiff, G.M.; Bernstein, D.I. Oral ribavirin treatment of influenza A and B. Antimicrob. Agents Chemother. 1987, 31, 1285–1287. [Google Scholar] [CrossRef]
- Turner, T.L.; Kopp, B.T.; Paul, G.; Landgrave, L.C.; Hayes, D., Jr.; Thompson, R. Respiratory syncytial virus: Current and emerging treatment options. ClinicoEcon. Outcomes Res. 2014, 6, 217–225. [Google Scholar] [CrossRef]
- Gavin, P.J.; Katz, B.Z. Intravenous ribavirin treatment for severe adenovirus disease in immunocompromised children. Pediatrics 2002, 110, e9. [Google Scholar] [CrossRef] [PubMed]
- McArdle, J.; Schafer, X.L.; Munger, J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J. Virol. 2011, 85, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.M.; Schafer, X.L.; Moorman, N.J.; Munger, J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J. Virol. 2011, 85, 5814–5824. [Google Scholar] [CrossRef] [PubMed]
- Courtney, R.J.; Steiner, S.M.; Benyesh-Melnick, M. Effects of 2-deoxy-D-glucose on herpes simplex virus replication. Virology 1973, 52, 447–455. [Google Scholar] [CrossRef]
- Passalacqua, K.D.; Lu, J.; Goodfellow, I.; Kolawole, A.O.; Arche, J.R.; Maddox, R.J.; Carnahan, K.E.; O’Riordan, M.X.D.; Wobus, C.E. Glycolysis Is an Intrinsic Factor for Optimal Replication of a Norovirus. mBio 2019, 10, e02175-18. [Google Scholar] [CrossRef]
- Namazue, J.; Kato, T.; Okuno, T.; Shiraki, K.; Yamanishi, K. Evidence for attachment of fatty acid to varicella-zoster virus glycoproteins and effect of cerulenin on the maturation of varicella-zoster virus glycoproteins. Intervirology 1989, 30, 268–277. [Google Scholar] [CrossRef]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [PubMed]
- Punjabi, A.; Traktman, P. Cell biological and functional characterization of the vaccinia virus F10 kinase: Implications for the mechanism of virion morphogenesis. J. Virol. 2005, 79, 2171–2190. [Google Scholar] [CrossRef]
- Greseth, M.D.; Traktman, P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 2014, 10, e1004021. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; George, D.; Prior, A.M.; Prasain, K.; Hao, S.; Le, D.D.; Hua, D.H.; Chang, K.O. Novel triacsin C analogs as potential antivirals against rotavirus infections. Eur. J. Med. Chem. 2012, 50, 311–318. [Google Scholar] [CrossRef]
- Guinea, R.; Carrasco, L. Phospholipid biosynthesis and poliovirus genome replication, two coupled phenomena. EMBO J. 1990, 9, 2011–2016. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, R.; Kumar, D.; Dube, S.N.; Singh, R.; Pandey, K.S.; Bag, B.C.; Kaushik, M.P.; Sekhar, K.; Dwarakanath, B.S.; Ravindranath, T. Acute toxicity and cardio-respiratory effects of 2-deoxy-D-glucose: A promising radio sensitiser. Biomed. Environ. Sci. 2006, 19, 96–103. [Google Scholar]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1958–1969. [Google Scholar] [CrossRef]
- Kovarik, J.J.; Kernbauer, E.; Holzl, M.A.; Hofer, J.; Gualdoni, G.A.; Schmetterer, K.G.; Miftari, F.; Sobanov, Y.; Meshcheryakova, A.; Mechtcheriakova, D.; et al. Fasting metabolism modulates the interleukin-12/interleukin-10 cytokine axis. PLoS ONE 2017, 12, e0180900. [Google Scholar] [CrossRef]
- Wang, A.; Huen, S.C.; Luan, H.H.; Yu, S.; Zhang, C.; Gallezot, J.D.; Booth, C.J.; Medzhitov, R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 2016, 166, 1512–1525.e1512. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef]
- Laszlo, J.; Harlan, W.R.; Klein, R.F.; Kirshner, N.; Estes, E.H., Jr.; Bogdonoff, M.D. The effect of 2-deoxy-D-glucose infusions on lipid and carbohydrate metabolism in man. J. Clin. Investig. 1961, 40, 171–176. [Google Scholar] [CrossRef]
- Fazio, N.; Buzzoni, R.; Baudin, E.; Antonuzzo, L.; Hubner, R.A.; Lahner, H.; WW, D.E.H.; Raderer, M.; Teule, A.; Capdevila, J.; et al. A Phase II Study of BEZ235 in Patients with Everolimus-resistant, Advanced Pancreatic Neuroendocrine Tumours. Anticancer Res. 2016, 36, 713–719. [Google Scholar] [PubMed]
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klumpen, H.J.; Capdevila, J.; et al. Phase II Study of BEZ235 versus Everolimus in Patients with Mammalian Target of Rapamycin Inhibitor-Naive Advanced Pancreatic Neuroendocrine Tumors. Oncologist 2018, 23, e766–e790. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Teo, G.; Bernardo, P.; Quinn, D.; Russell, K.; Klickstein, L.; Marshall, W.; Shergill, S. Targeting the biology of ageing with mTOR inhibitors to improve immune function in older adults: Phase 2b and phase 3 randomised trials. Lancet Healthy Longev. 2021, 2, e250–e262. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S.; Weiss, R.A. Human oncogenic viruses: Nature and discovery. Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 20160264. [Google Scholar] [CrossRef]
- Mui, U.N.; Haley, C.T.; Tyring, S.K. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. [Google Scholar] [CrossRef]
- Garber, K. Energy boost: The Warburg effect returns in a new theory of cancer. J. Natl. Cancer Inst. 2004, 96, 1805–1806. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleinehr, J.; Wilden, J.J.; Boergeling, Y.; Ludwig, S.; Hrincius, E.R. Metabolic Modifications by Common Respiratory Viruses and Their Potential as New Antiviral Targets. Viruses 2021, 13, 2068. https://doi.org/10.3390/v13102068
Kleinehr J, Wilden JJ, Boergeling Y, Ludwig S, Hrincius ER. Metabolic Modifications by Common Respiratory Viruses and Their Potential as New Antiviral Targets. Viruses. 2021; 13(10):2068. https://doi.org/10.3390/v13102068
Chicago/Turabian StyleKleinehr, Jens, Janine J. Wilden, Yvonne Boergeling, Stephan Ludwig, and Eike R. Hrincius. 2021. "Metabolic Modifications by Common Respiratory Viruses and Their Potential as New Antiviral Targets" Viruses 13, no. 10: 2068. https://doi.org/10.3390/v13102068
APA StyleKleinehr, J., Wilden, J. J., Boergeling, Y., Ludwig, S., & Hrincius, E. R. (2021). Metabolic Modifications by Common Respiratory Viruses and Their Potential as New Antiviral Targets. Viruses, 13(10), 2068. https://doi.org/10.3390/v13102068