
HSV-1 DNA Replication—Coordinated Regulation by Viral and Cellular Factors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Viral DNA Replication
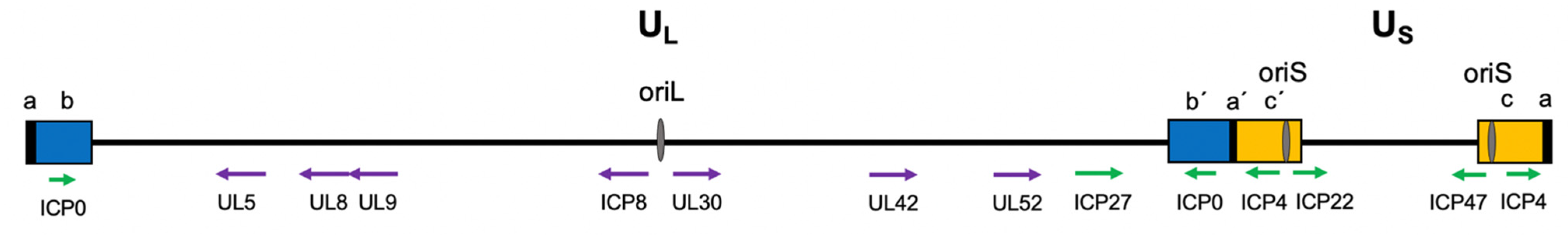
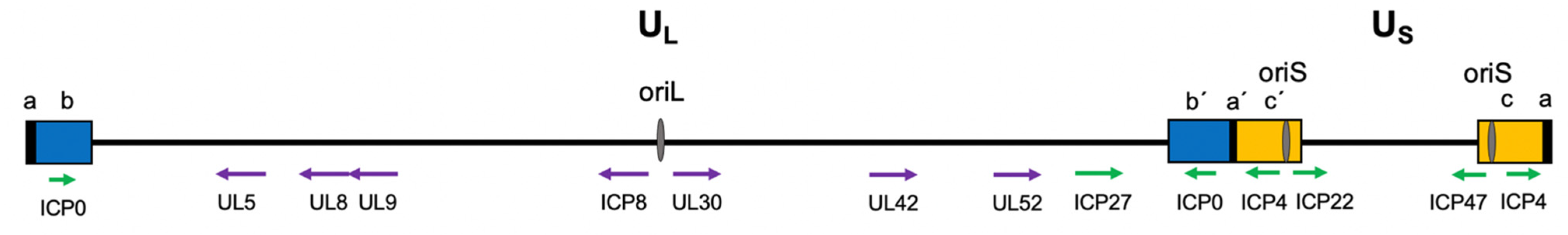
2.1. Viral Genome Structure
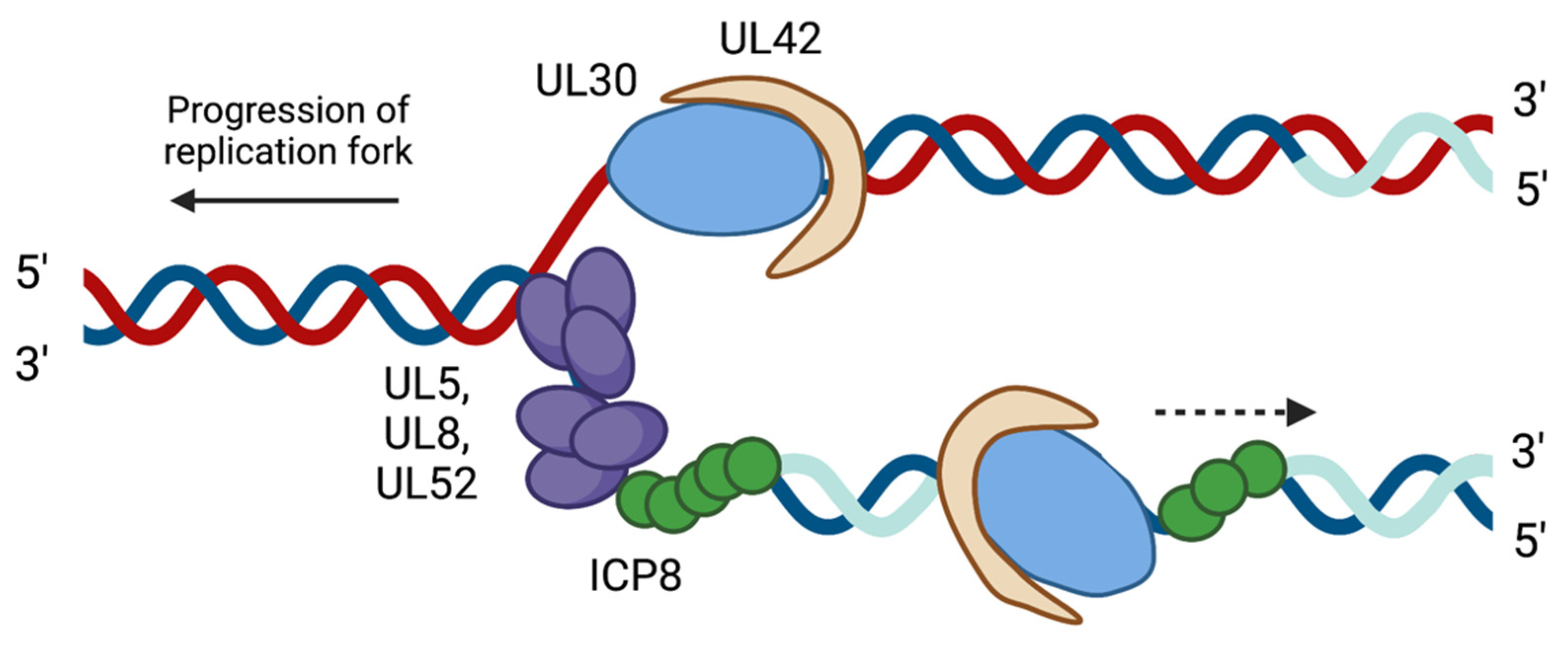
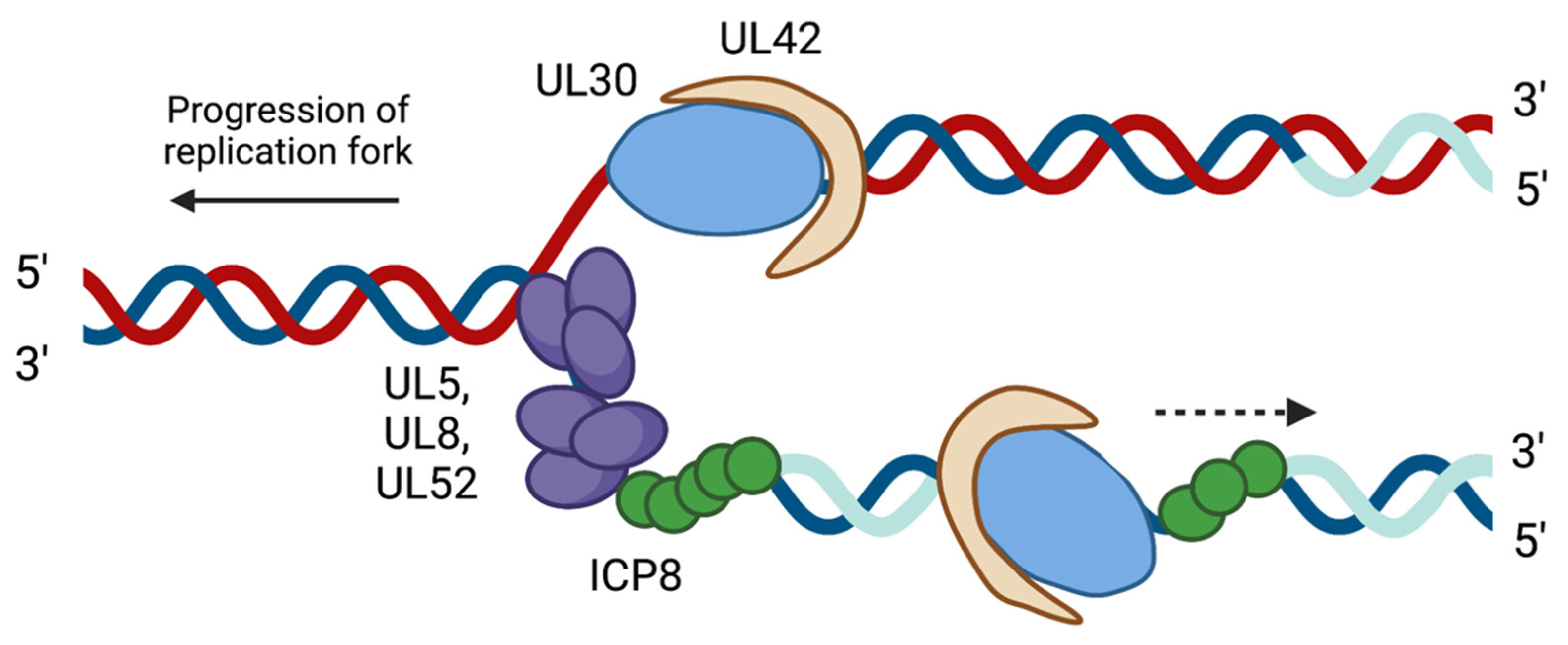
2.2. HSV-1 Replication Proteins
3. Replication-Coupled Processes
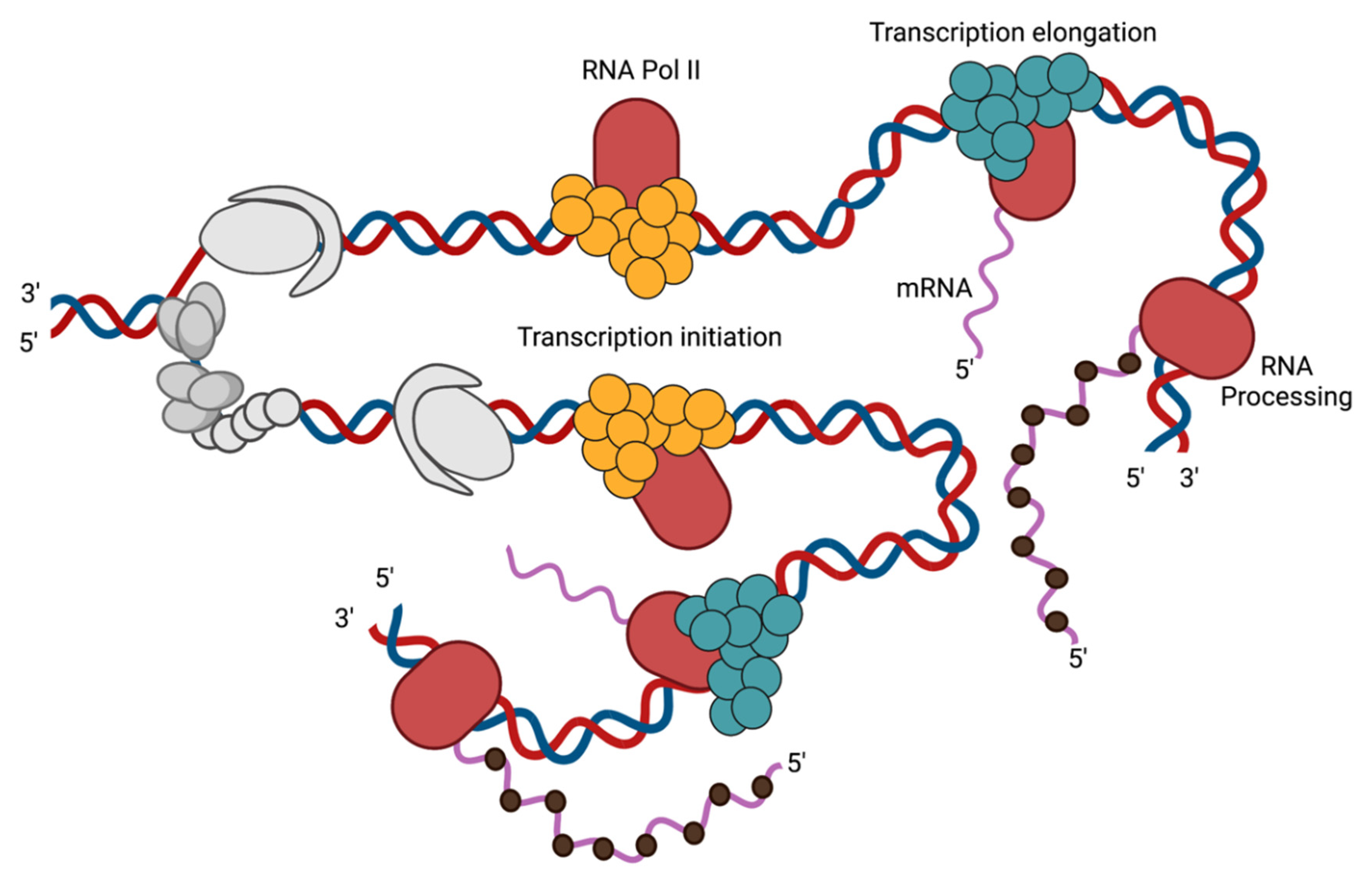
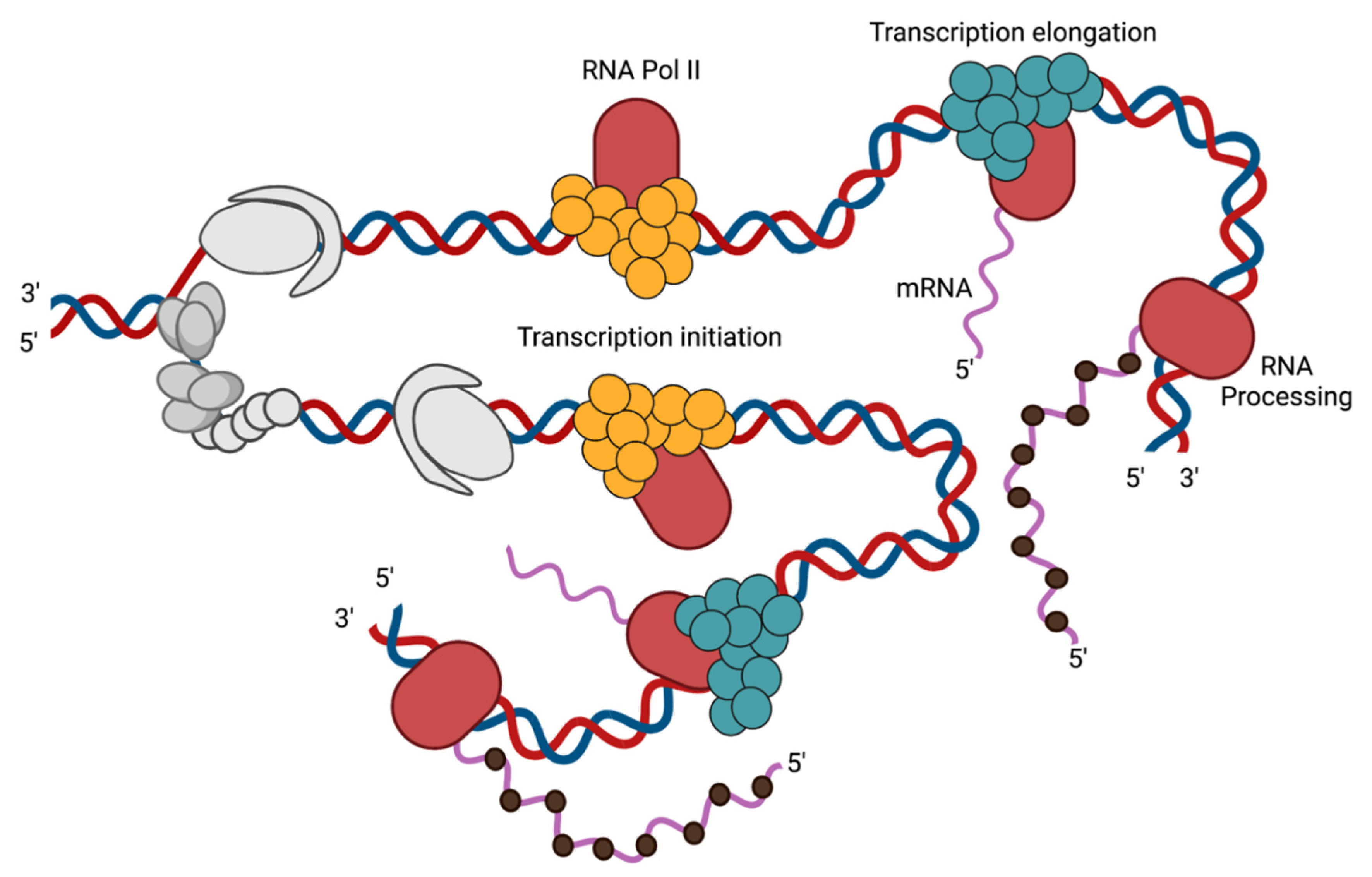
3.1. Transcription
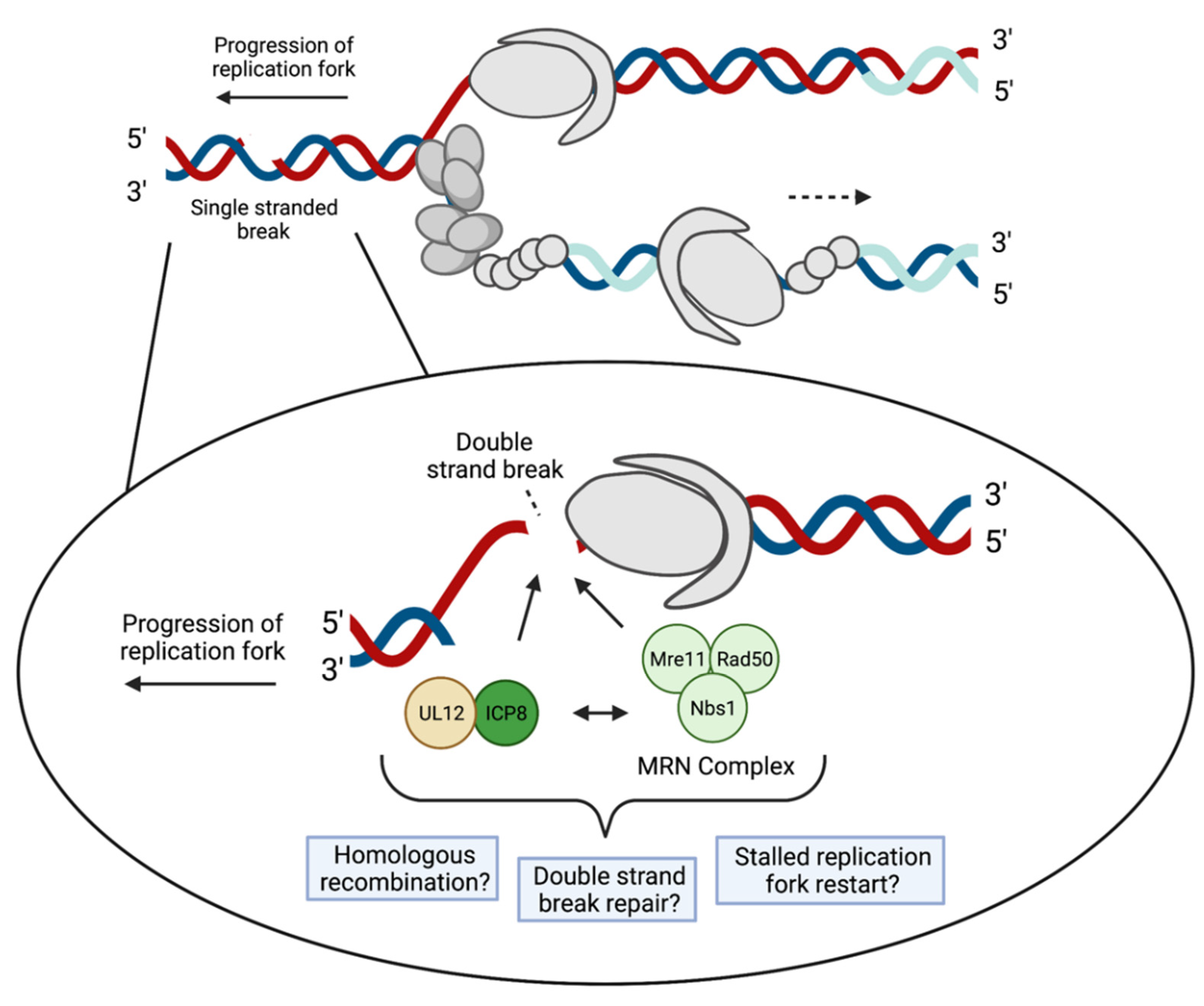
3.2. Recombination and Repair
3.3. Genome Packaging into Capsids
4. Viral Replication Fork Dynamics of Cellular Factors
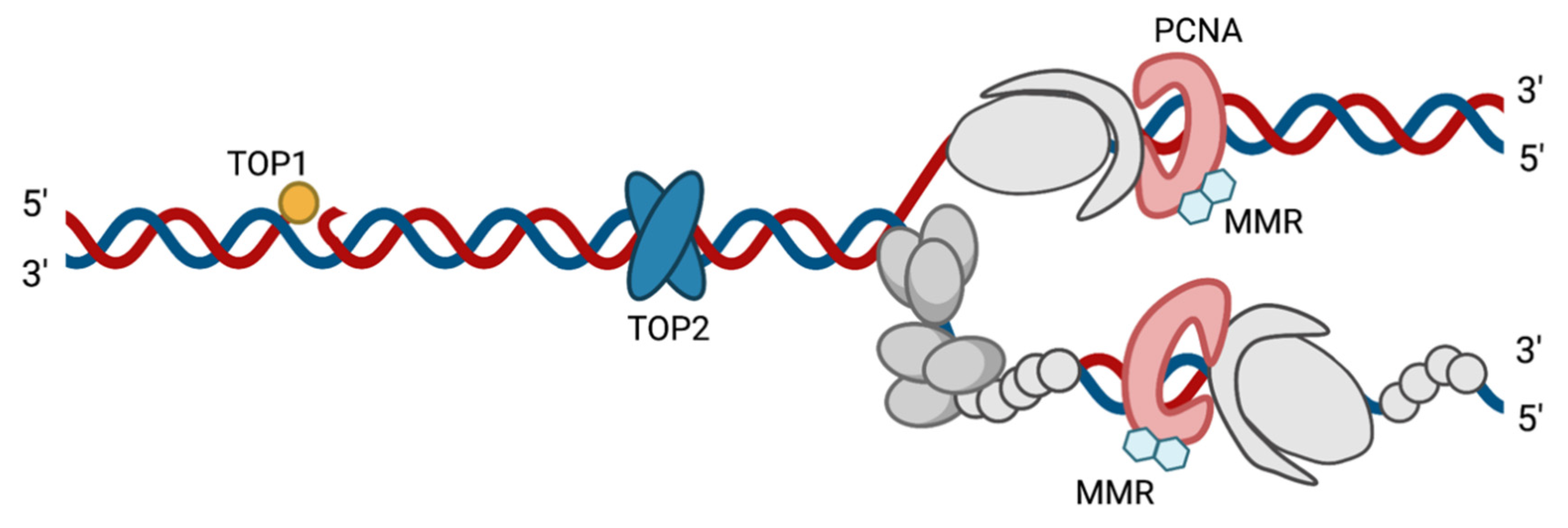
4.1. PCNA and MMR Proteins
4.2. Topoisomerases
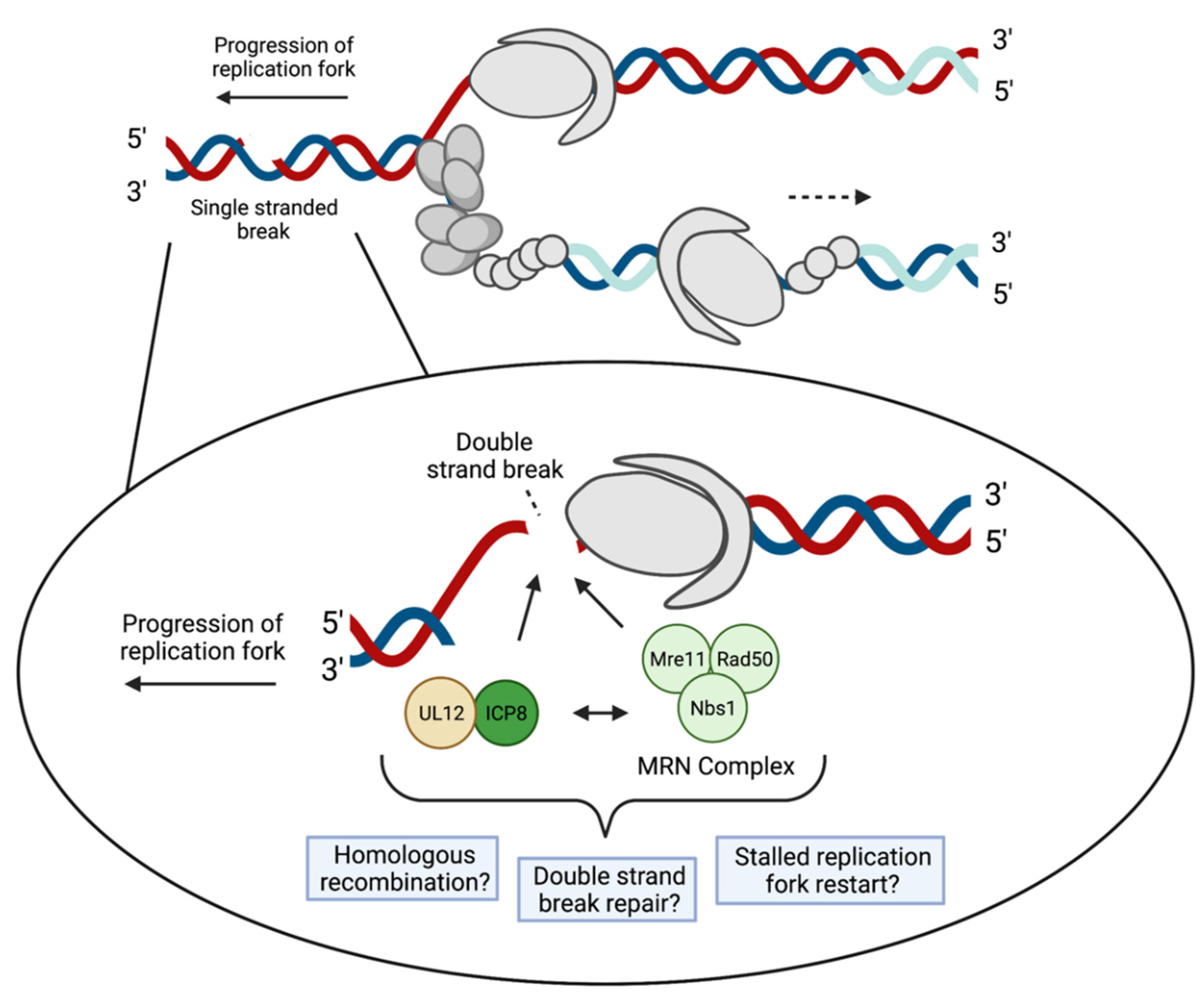
4.3. DNA Damage Response and DSB Repair Proteins
4.4. Transcription Factors
4.5. Chromatin Remodeling Factors
5. Remaining Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rechenchoski, D.Z.; Faccin-Galhardi, L.C.; Linhares, R.E.C.; Nozawa, C. Herpesvirus: An underestimated virus. Folia Microbiol. 2017, 62, 151–156. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.; Baines, J. Clinical management of herpes simplex virus infections: Past, present, and future. F1000Res 2018, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Alsweed, A.; Alsuhibani, M.; Casanova, J.L.; Al-Hajjar, S. Approach to recurrent Herpes Simplex Encephalitis in children. Int. J. Pediatr. Adolesc. Med. 2018, 5, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Jansen, M.; Haase, I.; Knebel-Mörsdorf, D. Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells. J. Gen. Virol. 2003, 84, 2473–2484. [Google Scholar] [CrossRef]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 2019, 10, 754. [Google Scholar] [CrossRef] [Green Version]
- Tombácz, D.; Csabai, Z.; Szűcs, A.; Balázs, Z.; Moldován, N.; Sharon, D.; Snyder, M.; Boldogkői, Z. Long-Read Isoform Sequencing Reveals a Hidden Complexity of the Transcriptional Landscape of Herpes Simplex Virus Type 1. Front. Microbiol. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Alwine, J.C.; Steinhart, W.L.; Hill, C.W. Transcription of herpes simplex type 1 DNA in nuclei isolated from infected HEp-2 and KB cells. Virology 1974, 60, 302–307. [Google Scholar] [CrossRef]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis: Sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. USA 1975, 72, 1276–1280. [Google Scholar] [CrossRef] [Green Version]
- Roizman, B.; Sears, A.E. An inquiry into the mechanisms of herpes simplex virus latency. Annu. Rev. Microbiol. 1987, 41, 543–571. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Wilson, A.C. Restarting Lytic Gene Transcription at the Onset of Herpes Simplex Virus Reactivation. J. Virol. 2017, 91, e01419-16. [Google Scholar] [CrossRef] [Green Version]
- Weller, S.K.; Coen, D.M. Herpes simplex viruses: Mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dembowski, J.A.; DeLuca, N.A. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 2015, 11, e1004939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dembowski, J.A.; Dremel, S.E.; DeLuca, N.A. Replication-Coupled Recruitment of Viral and Cellular Factors to Herpes Simplex Virus Type 1 Replication Forks for the Maintenance and Expression of Viral Genomes. PLoS Pathog. 2017, 13, e1006166. [Google Scholar] [CrossRef]
- Reyes, E.D.; Kulej, K.; Pancholi, N.J.; Akhtar, L.N.; Avgousti, D.C.; Kim, E.T.; Bricker, D.K.; Spruce, L.A.; Koniski, S.A.; Seeholzer, S.H.; et al. Identifying Host Factors Associated with DNA Replicated During Virus Infection. Mol. Cell Proteomics 2017, 16, 2079–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.A.; DeLuca, N.A. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. USA 2003, 100, 7871–7876. [Google Scholar] [CrossRef] [Green Version]
- Severini, A.; Scraba, D.G.; Tyrrell, D.L. Branched structures in the intracellular DNA of herpes simplex virus type 1. J. Virol. 1996, 70, 3169–3175. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porat, T.; Rixon, F.J. Replication of herpesvirus DNA IV: Analysis of concaterners. Virology 1979, 94, 61–70. [Google Scholar] [CrossRef]
- Smith, S.; Reuven, N.; Mohni, K.N.; Schumacher, A.J.; Weller, S.K. Structure of the herpes simplex virus 1 genome: Manipulation of nicks and gaps can abrogate infectivity and alter the cellular DNA damage response. J. Virol. 2014, 88, 10146–10156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Dremel, S.E.; DeLuca, N.A. Herpes simplex viral nucleoprotein creates a competitive transcriptional environment facilitating robust viral transcription and host shut off. eLife 2019, 8, e51109. [Google Scholar] [CrossRef]
- McSwiggen, D.T.; Hansen, A.S.; Teves, S.S.; Marie-Nelly, H.; Hao, Y.; Heckert, A.B.; Umemoto, K.K.; Dugast-Darzacq, C.; Tjian, R.; Darzacq, X. Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. eLife 2019, 8, e47098. [Google Scholar] [CrossRef]
- Oh, J.; Fraser, N.W. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 2008, 82, 3530–3537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinbach, S.S.; Summers, W.C. The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J. Gen. Virol. 1980, 51, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Mouttet, M.E.; Guétard, D.; Béchet, J.M. Random cleavage of intranuclear herpes simplex virus DNA by micrococcal nuclease. FEBS Lett. 1979, 100, 107–109. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Depledge, D.P.; Flores Cortes, E.; Breuer, J.; Schang, L.M. Chromatin dynamics and the transcriptional competence of HSV-1 genomes during lytic infections. PLoS Pathog. 2019, 15, e1008076. [Google Scholar] [CrossRef]
- Lacasse, J.J.; Schang, L.M. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J. Virol. 2010, 84, 1920–1933. [Google Scholar] [CrossRef] [Green Version]
- Challberg, M.D.; Kelly, T.J. Animal virus DNA replication. Annu. Rev. Biochem. 1989, 58, 671–717. [Google Scholar] [CrossRef]
- Martin, D.W.; Deb, S.P.; Klauer, J.S.; Deb, S. Analysis of the herpes simplex virus type 1 OriS sequence: Mapping of functional domains. J. Virol. 1991, 65, 4359–4369. [Google Scholar] [CrossRef] [Green Version]
- Summers, B.C.; Leib, D.A. Herpes simplex virus type 1 origins of DNA replication play no role in the regulation of flanking promoters. J. Virol. 2002, 76, 7020–7029. [Google Scholar] [CrossRef] [Green Version]
- Balliet, J.W.; Schaffer, P.A. Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. J. Virol. 2006, 80, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, I.R.; Boehmer, P.E. Replication of herpes simplex virus DNA. J. Biol. Chem. 1999, 274, 28059–28062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruckner, R.C.; Crute, J.J.; Dodson, M.S.; Lehman, I.R. The herpes simplex virus 1 origin binding protein: A DNA helicase. J. Biol. Chem. 1991, 266, 2669–2674. [Google Scholar] [CrossRef]
- Fierer, D.S.; Challberg, M.D. Purification and characterization of UL9, the herpes simplex virus type 1 origin-binding protein. J. Virol. 1992, 66, 3986–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.S.; Lehman, I.R. The herpes simplex virus type I origin binding protein. DNA-dependent nucleoside triphosphatase activity. J. Biol. Chem. 1993, 268, 1213–1219. [Google Scholar] [CrossRef]
- Boehmer, P.E.; Dodson, M.S.; Lehman, I.R. The herpes simplex virus type-1 origin binding protein. DNA helicase activity. J. Biol. Chem. 1993, 268, 1220–1225. [Google Scholar] [CrossRef]
- Martinez, R.; Shao, L.; Weller, S.K. The conserved helicase motifs of the herpes simplex virus type 1 origin-binding protein UL9 are important for function. J. Virol. 1992, 66, 6735–6746. [Google Scholar] [CrossRef] [Green Version]
- Weerasooriya, S.; DiScipio, K.A.; Darwish, A.S.; Bai, P.; Weller, S.K. Herpes simplex virus 1 ICP8 mutant lacking annealing activity is deficient for viral DNA replication. Proc. Natl. Acad. Sci. USA 2019, 116, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Tolun, G.; Makhov, A.M.; Ludtke, S.J.; Griffith, J.D. Details of ssDNA annealing revealed by an HSV-1 ICP8-ssDNA binary complex. Nucleic Acids Res. 2013, 41, 5927–5937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruyechan, W.T.; Weir, A.C. Interaction with nucleic acids and stimulation of the viral DNA polymerase by the herpes simplex virus type 1 major DNA-binding protein. J. Virol. 1984, 52, 727–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darwish, A.S.; Grady, L.M.; Bai, P.; Weller, S.K. ICP8 Filament Formation Is Essential for Replication Compartment Formation during Herpes Simplex Virus Infection. J. Virol. 2015, 90, 2561–2570. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, M.; Bushnell, D.A.; Elias, P.; Lehman, I.R. The UL8 subunit of the heterotrimeric herpes simplex virus type 1 helicase-primase is required for the unwinding of single strand DNA-binding protein (ICP8)-coated DNA substrates. J. Biol. Chem. 1997, 272, 22766–22770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, T.R.; Lehman, I.R. Functional interaction between the herpes simplex-1 DNA polymerase and UL42 protein. J. Biol. Chem. 1990, 265, 11227–11232. [Google Scholar] [CrossRef]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.E.; Elias, P.; Funnell, B.E.; Lehman, I.R. Interaction between the DNA polymerase and single-stranded DNA-binding protein (infected cell protein 8) of herpes simplex virus 1. J. Biol. Chem. 1987, 262, 4260–4266. [Google Scholar] [CrossRef]
- Crute, J.J.; Mocarski, E.S.; Lehman, I.R. A DNA helicase induced by herpes simplex virus type 1. Nucleic Acids Res. 1988, 16, 6585–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.S.; Crute, J.J.; Bruckner, R.C.; Lehman, I.R. Overexpression and assembly of the herpes simplex virus type 1 helicase-primase in insect cells. J. Biol. Chem. 1989, 264, 20835–20838. [Google Scholar] [CrossRef]
- Marintcheva, B.; Weller, S.K. A tale of two HSV-1 helicases: Roles of phage and animal virus helicases in DNA replication and recombination. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 77–118. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Chen, Y.; Weller, S.K. The two helicases of herpes simplex virus type 1 (HSV-1). Front. Biosci. 2006, 11, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, P.E.; Lehman, I.R. Herpes simplex virus DNA replication. Annu. Rev. Biochem. 1997, 66, 347–384. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Bai, P.; Mackay, S.; Korza, G.; Carson, J.H.; Kuchta, R.D.; Weller, S.K. Herpes simplex virus type 1 helicase-primase: DNA binding and consequent protein oligomerization and primase activation. J. Virol. 2011, 85, 968–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.P.; Du, W.J.; Huang, L.P.; Wei, Y.W.; Wu, H.L.; Feng, L.; Liu, C.M. The Pseudorabies Virus DNA Polymerase Accessory Subunit UL42 Directs Nuclear Transport of the Holoenzyme. Front. Microbiol. 2016, 7, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsden, H.S.; McLean, G.W.; Barnard, E.C.; Francis, G.J.; MacEachran, K.; Murphy, M.; McVey, G.; Cross, A.; Abbotts, A.P.; Stow, N.D. The catalytic subunit of the DNA polymerase of herpes simplex virus type 1 interacts specifically with the C terminus of the UL8 component of the viral helicase-primase complex. J. Virol. 1997, 71, 6390–6397. [Google Scholar] [CrossRef] [Green Version]
- Tsurumi, T.; Maeno, K.; Nishiyama, Y. Nucleotide sequence of the DNA polymerase gene of herpes simplex virus type 2 and comparison with the type 1 counterpart. Gene 1987, 52, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, K.; Piret, J.; Boivin, G. Herpesvirus DNA polymerases: Structures, functions and inhibitors. Virus Res. 2017, 234, 177–192. [Google Scholar] [CrossRef]
- Chaudhuri, M.; Song, L.; Parris, D.S. The herpes simplex virus type 1 DNA polymerase processivity factor increases fidelity without altering pre-steady-state rate constants for polymerization or excision. J. Biol. Chem. 2003, 278, 8996–9004. [Google Scholar] [CrossRef] [Green Version]
- Weisshart, K.; Chow, C.S.; Coen, D.M. Herpes simplex virus processivity factor UL42 imparts increased DNA-binding specificity to the viral DNA polymerase and decreased dissociation from primer-template without reducing the elongation rate. J. Virol. 1999, 73, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Bermek, O.; Willcox, S.; Griffith, J.D. DNA replication catalyzed by herpes simplex virus type 1 proteins reveals trombone loops at the fork. J. Biol. Chem. 2015, 290, 2539–2545. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, J.; Marcy, A.I.; Coen, D.M.; Challberg, M.D. The herpes simplex virus type 1 UL42 gene product: A subunit of DNA polymerase that functions to increase processivity. J. Virol. 1990, 64, 5976–5987. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.A.; Best, M.G.; Friedmann, T.; Parris, D.S. Isolation of a herpes simplex virus type 1 mutant deleted for the essential UL42 gene and characterization of its null phenotype. J. Virol. 1991, 65, 700–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parris, D.S.; Cross, A.; Haarr, L.; Orr, A.; Frame, M.C.; Murphy, M.; McGeoch, D.J.; Marsden, H.S. Identification of the gene encoding the 65-kilodalton DNA-binding protein of herpes simplex virus type 1. J. Virol. 1988, 62, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, M.; Parris, D.S. Evidence against a simple tethering model for enhancement of herpes simplex virus DNA polymerase processivity by accessory protein UL42. J. Virol. 2002, 76, 10270–10281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komazin-Meredith, G.; Santos, W.L.; Filman, D.J.; Hogle, J.M.; Verdine, G.L.; Coen, D.M. The positively charged surface of herpes simplex virus UL42 mediates DNA binding. J. Biol. Chem. 2008, 283, 6154–6161. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.S.; Coen, D.M. Mutations that specifically impair the DNA binding activity of the herpes simplex virus protein UL42. J. Virol. 1995, 69, 6965–6971. [Google Scholar] [CrossRef] [Green Version]
- Grady, L.M.; Szczepaniak, R.; Murelli, R.P.; Masaoka, T.; Le Grice, S.F.J.; Wright, D.L.; Weller, S.K. The Exonuclease Activity of Herpes Simplex Virus 1 UL12 Is Required for Production of Viral DNA That Can Be Packaged To Produce Infectious Virus. J. Virol. 2017, 91, e01380-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. “Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review. Viruses 2020, 13, 17. [Google Scholar] [CrossRef]
- Fyfe, J.A.; Keller, P.M.; Furman, P.A.; Miller, R.L.; Elion, G.B. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem. 1978, 253, 8721–8727. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Summers, W.C. Site-directed mutagenesis of a nucleotide-binding domain in HSV-1 thymidine kinase: Effects on catalytic activity. Virology 1988, 163, 638–642. [Google Scholar] [CrossRef]
- Batterson, W.; Roizman, B. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 1983, 46, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.E.; Palfreyman, J.W.; Preston, C.M. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 1984, 180, 1–19. [Google Scholar] [CrossRef]
- Watson, R.J.; Clements, J.B. A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature 1980, 285, 329–330. [Google Scholar] [CrossRef]
- O’Hare, P. The virion transactivator of herpes simplex virus. In Seminars in Virology; Elsevier: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Smith, C.A.; Bates, P.; Rivera-Gonzalez, R.; Gu, B.; DeLuca, N.A. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol. 1993, 67, 4676–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, J.T.; DeLuca, N.A. Herpes Simplex Virus 1 ICP4 Forms Complexes with TFIID and Mediator in Virus-Infected Cells. J. Virol. 2011, 85, 5733–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, P.; Deluca, N.A. Binding of ICP4, TATA-binding protein, and RNA polymerase II to herpes simplex virus type 1 immediate-early, early, and late promoters in virus-infected cells. J. Virol. 2008, 82, 2339–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dembowski, J.A.; DeLuca, N.A. Temporal Viral Genome-Protein Interactions Define Distinct Stages of Productive Herpesviral Infection. mBio 2018, 9, e01182-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, M.C.; Dybas, J.M.; Hughes, J.; Weitzman, M.D.; Boutell, C. The HSV-1 ubiquitin ligase ICP0: Modifying the cellular proteome to promote infection. Virus Res. 2020, 285, 198015. [Google Scholar] [CrossRef] [PubMed]
- Sandri-Goldin, R.M.; Mendoza, G.E. A herpesvirus regulatory protein appears to act post-transcriptionally by affecting mRNA processing. Genes Dev. 1992, 6, 848–863. [Google Scholar] [CrossRef] [Green Version]
- McGregor, F.; Phelan, A.; Dunlop, J.; Clements, J.B. Regulation of herpes simplex virus poly (A) site usage and the action of immediate-early protein IE63 in the early-late switch. J. Virol. 1996, 70, 1931–1940. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hennig, T.; Whisnant, A.W.; Erhard, F.; Prusty, B.K.; Friedel, C.C.; Forouzmand, E.; Hu, W.; Erber, L.; Chen, Y.; et al. Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat. Commun. 2020, 11, 293. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.H.; Sciabica, K.S.; Sandri-Goldin, R.M. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J. Virol. 2002, 76, 12877–12889. [Google Scholar] [CrossRef] [Green Version]
- Fox, H.L.; Dembowski, J.A.; DeLuca, N.A. A Herpesviral Immediate Early Protein Promotes Transcription Elongation of Viral Transcripts. mBio 2017, 8, e00745-17. [Google Scholar] [CrossRef] [Green Version]
- Taylor, T.J.; McNamee, E.E.; Day, C.; Knipe, D.M. Herpes simplex virus replication compartments can form by coalescence of smaller compartments. Virology 2003, 309, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.C.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. VIII. The transcription program consists of three phases during which both extent of transcription and accumulation of RNA in the cytoplasm are regulated. J. Virol. 1979, 31, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Dremel, S.E.; DeLuca, N.A. Genome replication affects transcription factor binding mediating the cascade of herpes simplex virus transcription. Proc. Natl. Acad. Sci. USA 2019, 116, 3734–3739. [Google Scholar] [CrossRef] [Green Version]
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell Biol. 2005, 25, 7158–7169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarisky, R.T.; Weber, P.C. Requirement for double-strand breaks but not for specific DNA sequences in herpes simplex virus type 1 genome isomerization events. J. Virol. 1994, 68, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, G.S.; Jacob, R.J.; Wadsworth, S.C.; Roizman, B. Anatomy of herpes simplex virus DNA: Evidence for four populations of molecules that differ in the relative orientations of their long and short components. Proc. Natl. Acad. Sci. USA 1975, 72, 4243–4247. [Google Scholar] [CrossRef] [Green Version]
- Mahiet, C.; Ergani, A.; Huot, N.; Alende, N.; Azough, A.; Salvaire, F.; Bensimon, A.; Conseiller, E.; Wain-Hobson, S.; Labetoulle, M.; et al. Structural variability of the herpes simplex virus 1 genome in vitro and in vivo. J. Virol. 2012, 86, 8592–8601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.; Weller, S.K. HSV-I and the cellular DNA damage response. Future Virol. 2015, 10, 383–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honess, R.W.; Buchan, A.; Halliburton, I.W.; Watson, D.H. Recombination and linkage between structural and regulatory genes of herpes simplex virus type 1: Study of the functional organization of the genome. J. Virol. 1980, 34, 716–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, L.S.; Buchman, T.G.; Roizman, B.; Schaffer, P.A. Anatomy of herpes simplex virus DNA. IX. Apparent exclusion of some parental DNA arrangements in the generation of intertypic (HSV-1 X HSV-2) recombinants. J. Virol. 1977, 24, 231–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, D.W.; Szpara, M.L. Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Burrel, S.; Boutolleau, D.; Ryu, D.; Agut, H.; Merkel, K.; Leendertz, F.H.; Calvignac-Spencer, S. Ancient Recombination Events between Human Herpes Simplex Viruses. Mol. Biol. Evol. 2017, 34, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Severini, A.; Morgan, A.R.; Tovell, D.R.; Tyrrell, D.L. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology 1994, 200, 428–435. [Google Scholar] [CrossRef]
- Jean, J.H.; Blankenship, M.L.; Ben-Porat, T. Replication of herpesvirus DNA. I. Electron microscopic analysis of replicative structures. Virology 1977, 79, 281–291. [Google Scholar] [CrossRef]
- Shlomai, J.; Friedmann, A.; Becker, Y. Replication intermediates of herpes simplex virus DNA. Virology 1976, 69, 647–659. [Google Scholar] [CrossRef]
- Martinez, R.; Sarisky, R.T.; Weber, P.C.; Weller, S.K. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J. Virol. 1996, 70, 2075–2085. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Efstathiou, S.; Simmons, A. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electrophoresis: Implications for viral DNA amplification strategies. Virology 1994, 202, 530–539. [Google Scholar] [CrossRef]
- Weller, S.K.; Sawitzke, J.A. Recombination promoted by DNA viruses: Phage λ to herpes simplex virus. Annu. Rev. Microbiol. 2014, 68, 237–258. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, A.J.; Mohni, K.N.; Kan, Y.; Hendrickson, E.A.; Stark, J.M.; Weller, S.K. The HSV-1 Exonuclease, UL12, Stimulates Recombination by a Single Strand Annealing Mechanism. PLoS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.B.; Staire, A.E.; Myers, R.S.; Weller, S.K. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 2003, 77, 7425–7433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuven, N.B.; Willcox, S.; Griffith, J.D.; Weller, S.K. Catalysis of strand exchange by the HSV-1 UL12 and ICP8 proteins: Potent ICP8 recombinase activity is revealed upon resection of dsDNA substrate by nuclease. J. Mol. Biol. 2004, 342, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [Green Version]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2011, 107, 1127–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, I.; Boyer, M.; Fraser, N.W. Early nucleosome deposition on, and replication of, HSV DNA requires cell factor PCNA. J. Neurovirol. 2015, 21, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Harland, J.; Dunn, P.; Cameron, E.; Conner, J.; Brown, S.M. The herpes simplex virus (HSV) protein ICP34.5 is a virion component that forms a DNA-binding complex with proliferating cell nuclear antigen and HSV replication proteins. J. Neurovirol. 2003, 9, 477–488. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Barnett, B.C. Neurovirulence factor. Nature 1991, 353, 609. [Google Scholar] [CrossRef]
- Mohni, K.N.; Mastrocola, A.S.; Bai, P.; Weller, S.K.; Heinen, C.D. DNA Mismatch Repair Proteins Are Required for Efficient Herpes Simplex Virus 1 Replication. J. Virol. 2011, 85, 12241. [Google Scholar] [CrossRef] [Green Version]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 2016, 39, 231–254. [Google Scholar] [CrossRef] [Green Version]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [Green Version]
- Hombauer, H.; Srivatsan, A.; Putnam, C.D.; Kolodner, R.D. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 2011, 334, 1713–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Contreras, A.J.; Ruppen, I.; Nieto-Soler, M.; Murga, M.; Rodriguez-Acebes, S.; Remeseiro, S.; Rodrigo-Perez, S.; Rojas, A.M.; Mendez, J.; Muñoz, J.; et al. A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 2013, 3, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, B.M.; McDonald, W.H.; Dungrawala, H.; Badu-Nkansah, A.; Kavanaugh, G.M.; Chen, Y.; Tabb, D.L.; Cortez, D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013, 288, 31458–31467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masih, P.J.; Kunnev, D.; Melendy, T. Mismatch Repair proteins are recruited to replicating DNA through interaction with Proliferating Cell Nuclear Antigen (PCNA). Nucleic Acids Res. 2008, 36, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelbrock, M.A.; Kaliyaperumal, S.; Williams, K.J. Structural, molecular and cellular functions of MSH2 and MSH6 during DNA mismatch repair, damage signaling and other noncanonical activities. Mutat. Res. 2013, 743-744, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Mohni, K.N.; Dee, A.R.; Smith, S.; Schumacher, A.J.; Weller, S.K. Efficient herpes simplex virus 1 replication requires cellular ATR pathway proteins. J. Virol. 2013, 87, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Karttunen, H.; Savas, J.N.; McKinney, C.; Chen, Y.H.; Yates, J.R., 3rd; Hukkanen, V.; Huang, T.T.; Mohr, I. Co-opting the Fanconi anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth. Mol. Cell 2014, 55, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Ebert, S.N.; Shtrom, S.S.; Muller, M.T. Topoisomerase II cleavage of herpes simplex virus type 1 DNA in vivo is replication dependent. J. Virol. 1990, 64, 4059–4066. [Google Scholar] [CrossRef] [Green Version]
- Francke, B.; Margolin, J. Effect of novobiocin and other DNA gyrase inhibitors on virus replication and DNA synthesis in herpes simplex virus type 1-infected BHK cells. J. Gen. Virol. 1981, 52, 401–404. [Google Scholar] [CrossRef]
- Advani, S.J.; Weichselbaum, R.R.; Roizman, B. Herpes simplex virus 1 activates cdc2 to recruit topoisomerase II alpha for post-DNA synthesis expression of late genes. Proc. Natl. Acad. Sci. USA 2003, 100, 4825–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Yamamoto, N.; Maeno, K.; Nishiyama, Y. Role of DNA topoisomerase I in the replication of herpes simplex virus type 2. Arch. Virol. 1990, 110, 121–127. [Google Scholar] [CrossRef]
- Bogani, F.; Chua, C.N.; Boehmer, P.E. Reconstitution of uracil DNA glycosylase-initiated base excision repair in herpes simplex virus-1. J. Biol. Chem. 2009, 284, 16784–16790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees-Miller, S.P.; Long, M.C.; Kilvert, M.A.; Lam, V.; Rice, S.A.; Spencer, C.A. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 1996, 70, 7471–7477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [Green Version]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, N.; Bai, P.; Buchek, G.; Korza, G.; Weller, S.K. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J. Virol. 2010, 84, 12504–12514. [Google Scholar] [CrossRef] [Green Version]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Mohni, K.N.; Livingston, C.M.; Cortez, D.; Weller, S.K. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J. Virol. 2010, 84, 12152–12164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Harancher, M.R.; Packard, J.E.; Cowan, S.P.; DeLuca, N.A.; Dembowski, J.A. Antiviral Properties of the LSD1 Inhibitor SP-2509. J. Virol. 2020, 94, e00974-20. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; DeLuca, N.A. Interaction of the viral activator protein ICP4 with TFIID through TAF250. Mol. Cell Biol. 1996, 16, 3085–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, L.M.; Bayer, A.; Deluca, N.A. Requirement of the N-terminal activation domain of herpes simplex virus ICP4 for viral gene expression. J. Virol. 2013, 87, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Wagner, L.M.; DeLuca, N.A. Temporal association of herpes simplex virus ICP4 with cellular complexes functioning at multiple steps in PolII transcription. PLoS ONE 2013, 8, e78242. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.M.; Kobiler, O. Gene Expression Correlates with the Number of Herpes Viral Genomes Initiating Infection in Single Cells. PLoS Pathog. 2016, 12, e1006082. [Google Scholar] [CrossRef] [PubMed]
- Drayman, N.; Patel, P.; Vistain, L.; Tay, S. HSV-1 single-cell analysis reveals the activation of anti-viral and developmental programs in distinct sub-populations. eLife 2019, 8, e46339. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Packard, J.E.; Dembowski, J.A. HSV-1 DNA Replication—Coordinated Regulation by Viral and Cellular Factors. Viruses 2021, 13, 2015. https://doi.org/10.3390/v13102015
Packard JE, Dembowski JA. HSV-1 DNA Replication—Coordinated Regulation by Viral and Cellular Factors. Viruses. 2021; 13(10):2015. https://doi.org/10.3390/v13102015
Chicago/Turabian StylePackard, Jessica E., and Jill A. Dembowski. 2021. "HSV-1 DNA Replication—Coordinated Regulation by Viral and Cellular Factors" Viruses 13, no. 10: 2015. https://doi.org/10.3390/v13102015
APA StylePackard, J. E., & Dembowski, J. A. (2021). HSV-1 DNA Replication—Coordinated Regulation by Viral and Cellular Factors. Viruses, 13(10), 2015. https://doi.org/10.3390/v13102015