
Embryonic Origins of Virus-Induced Hearing Loss: Overview of Molecular Etiology

{kind=link}
Abstract
1. Introduction
2. Initial Cellular Hallmarks of Auditory System Development
3. Molecular Determinants of Inner Ear Development
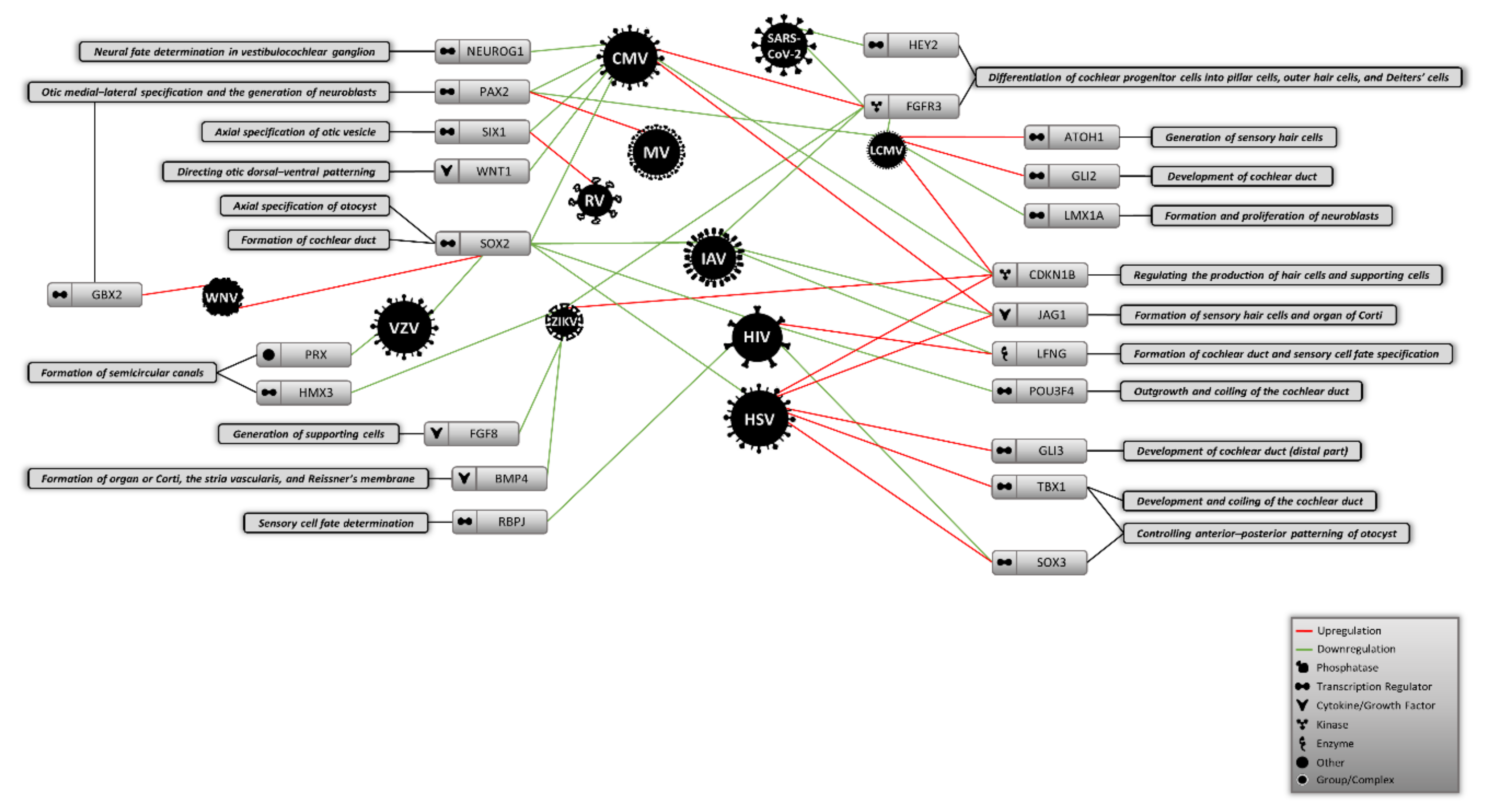
4. Potential Molecular Origins of Virus-Induced Hearing Loss
4.1. Cytomegalovirus
4.2. Herpes Simplex Virus
4.3. Rubella Virus
4.4. Lymphocytic Choriomeningitis Virus
4.5. Human Immunodeficiency Virus
4.6. Viruses Causing Acquired Hearing Loss
4.7. Other Viruses that Might Potentially Affect Inner Ear Development
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Elzouki, A.Y.; Harfi, H.A.; Nazer, H.; Oh, W.; Stapleton, F.; Whitley, R.J. Textbook of Clinical Pediatrics; Springer Science & Business Media: Berlin, Germany, 2011. [Google Scholar]
- Mazaheryazdi, M.; Aghasoleimani, M.; Karimi, M.; Arjmand, P. Perception of musical emotion in the students with cognitive and acquired hearing loss. Iran. J. Child Neurol. 2018, 12, 41. [Google Scholar] [PubMed]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Adler, S.P. Congenital cytomegalovirus screening. Pediatric Infect. Dis. J. 2005, 24, 1105–1106. [Google Scholar] [CrossRef] [PubMed]
- Bale, J.F., Jr. Cytomegalovirus Infections. Seminars in Pediatric Neurology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 101–106. [Google Scholar]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary hearing loss and deafness overview. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Fowler, K.B.; Dahle, A.J.; Boppana, S.B.; Pass, R.F. Newborn hearing screening: Will children with hearing loss caused by congenital cytomegalovirus infection be missed? J. Pediatrics 1999, 135, 60–64. [Google Scholar] [CrossRef]
- Fowler, K.B.; McCollister, F.P.; Dahle, A.J.; Boppana, S.; Britt, W.J.; Pass, R.F. Progressive and fluctuating sensorineural hearing loss in children with asymptomatic congenital cytomegalovirus infection. J. Pediatrics 1997, 130, 624–630. [Google Scholar] [CrossRef]
- Schraff, S.A.; Schleiss, M.R.; Brown, D.K.; Meinzen-Derr, J.; Choi, K.Y.; Greinwald, J.H.; Choo, D.I. Macrophage inflammatory proteins in cytomegalovirus-related inner ear injury. Otolaryngol.—Head Neck Surg. 2007, 137, 612–618. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Bowden, D.S. Rubella virus replication and links to teratogenicity. Clin. Microbiol. Rev. 2000, 13, 571–587. [Google Scholar] [CrossRef]
- Cohen, B.E.; Durstenfeld, A.; Roehm, P.C. Viral causes of hearing loss: A review for hearing health professionals. Trends Hear. 2014, 18, 2331216514541361. [Google Scholar] [CrossRef]
- Webster, W.S. Teratogen update: Congenital rubella. Teratology 1998, 58, 13–23. [Google Scholar] [CrossRef]
- Poppas, D.G., Jr.; Sekhar, H.K.C.; Lim, J.; Hillman, D.E. Ultrastructural findings in the cochlea of AIDS cases. Otol. Neurotol. 1994, 15, 456–465. [Google Scholar]
- Grimaldi, L.; Luzi, L.; Martino, G.; Furlan, R.; Nemni, R.; Antonelli, A.; Canal, N.; Pozza, G. Bilateral eighth cranial nerve neuropathy in human immunodeficiency virus infection. J. Neurol. 1993, 240, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Rarey, K.E. Otologic pathophysiology in patients with human immunodeficiency virus. Am. J. Otolaryngol. 1990, 11, 366–369. [Google Scholar] [CrossRef]
- Nomura, Y.; Kurata, T.; Saito, K. Cochlear changes after herpes simplex virus infection. Acta Oto-Laryngol. 1985, 99, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.I.; Parker, M.A.; Ryals, B.M.; Cotanche, D.A. Regeneration and replacement in the vertebrate inner ear. Drug Discov. Today 2005, 10, 1307–1312. [Google Scholar] [CrossRef]
- Goodrich, L.V. Early development of the spiral ganglion. In The Primary Auditory Neurons of the Mammalian Cochlea; Springer: New York, NY, USA, 2016; pp. 11–48. [Google Scholar]
- Torres, M.; Giráldez, F. The development of the vertebrate inner ear. Mech. Dev. 1998, 71, 5–21. [Google Scholar] [CrossRef]
- Fekete, D.M.; Wu, D.K. Revisiting cell fate specification in the inner ear. Curr. Opin. Neurobiol. 2002, 12, 35–42. [Google Scholar] [CrossRef]
- Durruthy-Durruthy, R.; Gottlieb, A.; Hartman, B.H.; Waldhaus, J.; Laske, R.D.; Altman, R.; Heller, S. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell 2014, 157, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Brigande, J.V.; Kiernan, A.E.; Gao, X.; Iten, L.E.; Fekete, D.M. Molecular genetics of pattern formation in the inner ear: Do compartment boundaries play a role? Proc. Natl. Acad. Sci. USA 2000, 97, 11700–11706. [Google Scholar] [CrossRef] [PubMed]
- Appler, J.M.; Goodrich, L.V. Connecting the ear to the brain: Molecular mechanisms of auditory circuit assembly. Prog. Neurobiol. 2011, 93, 488–508. [Google Scholar] [CrossRef]
- Cole, L.K.; Le Roux, I.; Nunes, F.; Laufer, E.; Lewis, J.; Wu, D.K. Sensory organ generation in the chicken inner ear: Contributions of bone morphogenetic protein 4, serrate1, and lunatic fringe. J. Comp. Neurol. 2000, 424, 509–520. [Google Scholar] [CrossRef]
- Zheng, W.; Huang, L.; Wei, Z.-B.; Silvius, D.; Tang, B.; Xu, P.-X. The role of Six1 in mammalian auditory system development. Development 2003, 130, 3989–4000. [Google Scholar] [CrossRef] [PubMed]
- Alsina, B.; Abelló, G.; Ulloa, E.; Henrique, D.; Pujades, C.; Giraldez, F. FGF signaling is required for determination of otic neuroblasts in the chick embryo. Dev. Biol. 2004, 267, 119–134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kiernan, A.E.; Pelling, A.L.; Leung, K.K.; Tang, A.S.; Bell, D.M.; Tease, C.; Lovell-Badge, R.; Steel, K.P.; Cheah, K.S. Sox2 is required for sensory organ development in the mammalian inner ear. Nature 2005, 434, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.K.; Kelley, M.W. Molecular mechanisms of inner ear development. Cold Spring Harb. Perspect. Biol. 2012, 4, a008409. [Google Scholar] [CrossRef]
- Reijntjes, S.; Gale, E.; Maden, M. Generating gradients of retinoic acid in the chick embryo: Cyp26C1 expression and a comparative analysis of the Cyp26 enzymes. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2004, 230, 509–517. [Google Scholar]
- Bok, J.; Bronner-Fraser, M.; Wu, D.K. Role of the hindbrain in dorsoventral but not anteroposterior axial specification of the inner ear. Development 2005, 132, 2115–2124. [Google Scholar] [CrossRef]
- Radosevic, M.; Robert-Moreno, À.; Coolen, M.; Bally-Cuif, L.; Alsina, B. Her9 represses neurogenic fate downstream of Tbx1 and retinoic acid signaling in the inner ear. Development 2011, 138, 397–408. [Google Scholar] [CrossRef]
- Abello, G.; Khatri, S.; Radosevic, M.; Scotting, P.; Giraldez, F.; Alsina, B. Independent regulation of Sox3 and Lmx1b by FGF and BMP signaling influences the neurogenic and non-neurogenic domains in the chick otic placode. Dev. Biol. 2010, 339, 166–178. [Google Scholar] [CrossRef]
- Brown, A.S.; Epstein, D.J. Otic ablation of smoothened reveals direct and indirect requirements for Hedgehog signaling in inner ear development. Development 2011, 138, 3967–3976. [Google Scholar] [CrossRef]
- Bok, J.; Dolson, D.K.; Hill, P.; Rüther, U.; Epstein, D.J.; Wu, D.K. Opposing gradients of Gli repressor and activators mediate Shh signaling along the dorsoventral axis of the inner ear. Development 2007, 134, 1713–1722. [Google Scholar] [CrossRef]
- Ohyama, T.; Mohamed, O.A.; Taketo, M.M.; Dufort, D.; Groves, A.K. Wnt signals mediate a fate decision between otic placode and epidermis. Development 2006, 133, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, C.S.; Ohyama, T.; Segil, N.; Groves, A.K. Notch signaling augments the canonical Wnt pathway to specify the size of the otic placode. Development 2008, 135, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Cantos, R.; Patente, M.; Wu, D.K. Gbx2 is required for the morphogenesis of the mouse inner ear: A downstream candidate of hindbrain signaling. Development 2005, 132, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Koundakjian, E.J.; Appler, J.L.; Goodrich, L.V. Auditory neurons make stereotyped wiring decisions before maturation of their targets. J. Neurosci. 2007, 27, 14078–14088. [Google Scholar] [CrossRef]
- Bell, D.; Streit, A.; Gorospe, I.; Varela-Nieto, I.; Alsina, B.; Giraldez, F. Spatial and temporal segregation of auditory and vestibular neurons in the otic placode. Dev. Biol. 2008, 322, 109–120. [Google Scholar] [CrossRef]
- Koo, S.K.; Hill, J.K.; Hwang, C.H.; Lin, Z.S.; Millen, K.J.; Wu, D.K. Lmx1a maintains proper neurogenic, sensory, and non-sensory domains in the mammalian inner ear. Dev. Biol. 2009, 333, 14–25. [Google Scholar] [CrossRef]
- Raft, S.; Koundakjian, E.J.; Quinones, H.; Jayasena, C.S.; Goodrich, L.V.; Johnson, J.E.; Segil, N.; Groves, A.K. Cross-regulation of Ngn1 and Math1 coordinates the production of neurons and sensory hair cells during inner ear development. Development 2007, 134, 4405–4415. [Google Scholar] [CrossRef]
- Freyer, L.; Morrow, B.E. Canonical Wnt signaling modulates Tbx1, Eya1, and Six1 expression, restricting neurogenesis in the otic vesicle. Dev. Dyn. 2010, 239, 1708–1722. [Google Scholar] [CrossRef]
- Brooker, R.; Hozumi, K.; Lewis, J. Notch ligands with contrasting functions: Jagged1 and Delta1 in the mouse inner ear. Development 2006, 133, 1277–1286. [Google Scholar] [CrossRef]
- Basch, M.L.; Ohyama, T.; Segil, N.; Groves, A.K. Canonical Notch signaling is not necessary for prosensory induction in the mouse cochlea: Insights from a conditional mutant of RBPjκ. J. Neurosci. 2011, 31, 8046–8058. [Google Scholar] [CrossRef]
- Yamamoto, N.; Chang, W.; Kelley, M.W. Rbpj regulates development of prosensory cells in the mammalian inner ear. Dev. Biol. 2011, 353, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Riccomagno, M.M.; Takada, S.; Epstein, D.J. Wnt-dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh. Genes Dev. 2005, 19, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Acampora, D.; Merlo, G.R.; Paleari, L.; Zerega, B.; Postiglione, M.P.; Mantero, S.; Bober, E.; Barbieri, O.; Simeone, A.; Levi, G. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development 1999, 126, 3795–3809. [Google Scholar] [PubMed]
- Depew, M.J.; Liu, J.K.; Long, J.E.; Presley, R.; Meneses, J.J.; Pedersen, R.A.; Rubenstein, J. Dlx5 regulates regional development of the branchial arches and sensory capsules. Development 1999, 126, 3831–3846. [Google Scholar]
- Merlo, G.R.; Paleari, L.; Mantero, S.; Zerega, B.; Adamska, M.; Rinkwitz, S.; Bober, E.; Levi, G. The Dlx5 homeobox gene is essential for vestibular morphogenesis in the mouse embryo through a BMP4-mediated pathway. Dev. Biol. 2002, 248, 157–169. [Google Scholar] [CrossRef]
- Hadrys, T.; Braun, T.; Rinkwitz-Brandt, S.; Arnold, H.-H.; Bober, E. Nkx5-1 controls semicircular canal formation in the mouse inner ear. Development 1998, 125, 33–39. [Google Scholar]
- Wang, W.; Van De Water, T.; Lufkin, T. Inner ear and maternal reproductive defects in mice lacking the Hmx3 homeobox gene. Development 1998, 125, 621–634. [Google Scholar]
- ten Berge, D.; Brouwer, A.; Korving, J.; Martin, J.F.; Meijlink, F. Prx1 and Prx2 in skeletogenesis: Roles in the craniofacial region, inner ear and limbs. Development 1998, 125, 3831–3842. [Google Scholar]
- Phippard, D.; Lu, L.; Lee, D.; Saunders, J.C.; Crenshaw, E.B. Targeted mutagenesis of the POU-domain GeneBrn4/Pou3f4 causes developmental defects in the inner ear. J. Neurosci. 1999, 19, 5980–5989. [Google Scholar] [CrossRef]
- Sobol, S.E.; Teng, X.; Crenshaw, E.B. Abnormal mesenchymal differentiation in the superior semicircular canal of brn4/pou3f4 knockout mice. Arch. Otolaryngol.–Head Neck Surg. 2005, 131, 41–45. [Google Scholar] [CrossRef]
- Ohyama, T.; Basch, M.L.; Mishina, Y.; Lyons, K.M.; Segil, N.; Groves, A.K. BMP signaling is necessary for patterning the sensory and nonsensory regions of the developing mammalian cochlea. J. Neurosci. 2010, 30, 15044–15051. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Segil, N. p27 (Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development 1999, 126, 1581–1590. [Google Scholar] [PubMed]
- Helms, A.W.; Abney, A.L.; Ben-Arie, N.; Zoghbi, H.Y.; Johnson, J.E. Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 2000, 127, 1185–1196. [Google Scholar] [PubMed]
- Anniko, M. Cytodifferentiation of cochlear hair cells. Am. J. Otolaryngol. 1983, 4, 375–388. [Google Scholar] [CrossRef]
- Mueller, K.L.; Jacques, B.E.; Kelley, M.W. Fibroblast growth factor signaling regulates pillar cell development in the organ of corti. J. Neurosci. 2002, 22, 9368–9377. [Google Scholar] [CrossRef]
- Jacques, B.E.; Montcouquiol, M.E.; Layman, E.M.; Lewandoski, M.; Kelley, M.W. Fgf8 induces pillar cell fate and regulates cellular patterning in the mammalian cochlea. Development 2007, 134, 3021–3029. [Google Scholar] [CrossRef]
- Woods, C.; Montcouquiol, M.; Kelley, M.W. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat. Neurosci. 2004, 7, 1310–1318. [Google Scholar] [CrossRef]
- Shim, K.; Minowada, G.; Coling, D.E.; Martin, G.R. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev. Cell 2005, 8, 553–564. [Google Scholar] [CrossRef]
- Doetzlhofer, A.; Basch, M.L.; Ohyama, T.; Gessler, M.; Groves, A.K.; Segil, N. Hey2 regulation by FGF provides a Notch-independent mechanism for maintaining pillar cell fate in the organ of Corti. Dev. Cell 2009, 16, 58–69. [Google Scholar] [CrossRef]
- Wu, C.-C.; Jiang, X.; Wang, X.-Z.; Liu, X.-J.; Li, X.-J.; Yang, B.; Ye, H.-Q.; Harwardt, T.; Jiang, M.; Xia, H.-M. Human cytomegalovirus immediate early 1 protein causes loss of SOX2 from neural progenitor cells by trapping unphosphorylated STAT3 in the nucleus. J. Virol. 2018, 92, e00340-18. [Google Scholar] [CrossRef]
- Browne, E.P.; Wing, B.; Coleman, D.; Shenk, T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: Viral block to the accumulation of antiviral mRNAs. J. Virol. 2001, 75, 12319–12330. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fang, F.; Dong, Y.; Zhou, H.; Zhen, H.; Liu, J.; Li, G. Inhibitory effect of murine cytomegalovirus infection on neural stem cells’ differentiation and its mechanisms. Zhonghua Er Ke Za Zhi= Chin. J. Pediatrics 2006, 44, 505–508. [Google Scholar]
- Thomas, K.R.; Capecchi, M.R. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature 1990, 346, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Smith, P.M.; Yurochko, A.D. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J. Immunol. 2008, 181, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, A.E.; Xu, J.; Gridley, T. The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet 2006, 2, e4. [Google Scholar] [CrossRef]
- Chen, Z.; Knutson, E.; Kurosky, A.; Albrecht, T. Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J. Virol. 2001, 75, 3613–3625. [Google Scholar] [CrossRef]
- Martínez, F.P.; Tang, Q. Identification of cellular proteins that interact with human cytomegalovirus immediate-early protein 1 by protein array assay. Viruses 2014, 6, 89–105. [Google Scholar] [CrossRef]
- Hsu, J.-L.; van den Boomen, D.J.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J. Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by HCMV US2 in cooperation with UL141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef]
- Hayashi, T.; Cunningham, D.; Bermingham-McDonogh, O. Loss of Fgfr3 leads to excess hair cell development in the mouse organ of Corti. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 525–533. [Google Scholar] [CrossRef]
- Puligilla, C.; Feng, F.; Ishikawa, K.; Bertuzzi, S.; Dabdoub, A.; Griffith, A.J.; Fritzsch, B.; Kelley, M.W. Disruption of fibroblast growth factor receptor 3 signaling results in defects in cellular differentiation, neuronal patterning, and hearing impairment. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 1905–1917. [Google Scholar]
- Kimberlin, D.W.; Lin, C.-Y.; Sánchez, P.J.; Demmler, G.J.; Dankner, W.; Shelton, M.; Jacobs, R.F.; Vaudry, W.; Pass, R.F.; Kiell, J.M. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: A randomized, controlled trial. J. Pediatrics 2003, 143, 16–25. [Google Scholar] [CrossRef]
- Shin, J.J.; Keamy, D.G., Jr.; Steinberg, E.A. Medical and surgical interventions for hearing loss associated with congenital cytomegalovirus: A systematic review. Otolaryngol.-Head Neck Surg. 2011, 144, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, S.; Morioka, I.; Fukushima, S.; Yamana, K.; Nishida, K.; Iwatani, S.; Fujioka, K.; Matsumoto, H.; Imanishi, T.; Nakamachi, Y. Efficacy of valganciclovir treatment depends on the severity of hearing dysfunction in symptomatic infants with congenital cytomegalovirus infection. Int. J. Mol. Sci. 2019, 20, 1388. [Google Scholar] [CrossRef]
- Al Muhaimeed, H.; Zakzouk, S.M. Hearing loss and herpes simplex. J. Trop. Pediatrics 1997, 43, 20–24. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lavi, E.S.; Sklar, E.M. Enhancement of the eighth cranial nerve and labyrinth on MR imaging in sudden sensorineural hearing loss associated with human herpesvirus 1 infection: Case report. Am. J. Neuroradiol. 2001, 22, 1380–1382. [Google Scholar]
- Mimura, T.; Amano, S.; Nagahara, M.; Oshika, T.; Tsushima, K.; Nakanishi, N.; Tanino, T. Corneal endotheliitis and idiopathic sudden sensorineural hearing loss. Am. J. Ophthalmol. 2002, 133, 699–700. [Google Scholar] [CrossRef]
- Rabinstein, A.; Jerry, J.; Saraf–Lavi, E.; Sklar, E.; Bradley, W. Sudden sensorineural hearing loss associated with herpes simplex virus type 1 infection. Neurology 2001, 56, 571–572. [Google Scholar] [CrossRef]
- Rotschafer, J.H.; Hu, S.; Little, M.; Erickson, M.; Low, W.C.; Cheeran, M.C. Modulation of neural stem/progenitor cell proliferation during experimental Herpes Simplex encephalitis is mediated by differential FGF-2 expression in the adult brain. Neurobiol. Dis. 2013, 58, 144–155. [Google Scholar] [CrossRef]
- Hu, B.; Li, X.; Huo, Y.; Yu, Y.; Zhang, Q.; Chen, G.; Zhang, Y.; Fraser, N.W.; Wu, D.; Zhou, J. Cellular responses to HSV-1 infection are linked to specific types of alterations in the host transcriptome. Sci. Rep. 2016, 6, 28075. [Google Scholar] [CrossRef]
- Sánchez-Quiles, V.; Mora, M.I.; Segura, V.; Greco, A.; Epstein, A.L.; Foschini, M.G.; Dayon, L.; Sanchez, J.-C.; Prieto, J.; Corrales, F.J. HSV-1 Cgal+ infection promotes quaking RNA binding protein production and induces nuclear-cytoplasmic shuttling of quaking I-5 isoform in human hepatoma cells. Mol. Cell. Proteom. 2011, 10, 6. [Google Scholar] [CrossRef]
- Derebery, M.J.; Fisher, L.M.; Iqbal, Z. Randomized double-blinded, placebo-controlled clinical trial of famciclovir for reduction of Ménière’s disease symptoms. Otolaryngol.—Head Neck Surg. 2004, 131, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.; Bauer, M.; Neumann, J.; Lüdde, A.; Rennert, P.; Friedrich, N.; Claus, C.; Perelygina, L.; Mankertz, A. Gene expression profiling of rubella virus infected primary endothelial cells of fetal and adult origin. Virol. J. 2016, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Nakamura, K.; Funahashi, J.-I.; Ikeda, K.; Yamada, G.; Tokano, H.; Okamura, H.-O.; Kitamura, K.; Muto, S.; Kotaki, H. Six1 controls patterning of the mouse otic vesicle. Development 2004, 131, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Bilz, N.C.; Willscher, E.; Binder, H.; Böhnke, J.; Stanifer, M.L.; Hübner, D.; Boulant, S.; Liebert, U.G.; Claus, C. Teratogenic rubella virus alters the endodermal differentiation capacity of human induced pluripotent stem cells. Cells 2019, 8, 870. [Google Scholar] [CrossRef]
- McLean, H.Q.; Fiebelkorn, A.P.; Temte, J.L.; Wallace, G.S. Prevention of measles, rubella, congenital rubella syndrome, and mumps, 2013: Summary recommendations of the Advisory Committee on Immunization Practices (ACIP). Morb. Mortal. Wkly. Recomm. Rep. 2013, 62, 1–34. [Google Scholar]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef]
- Parmigiani, E.; Leto, K.; Rolando, C.; Figueres-Onate, M.; López-Mascaraque, L.; Buffo, A.; Rossi, F. Heterogeneity and bipotency of astroglial-like cerebellar progenitors along the interneuron and glial lineages. J. Neurosci. 2015, 35, 7388–7402. [Google Scholar] [CrossRef]
- Klein de Licona, H.W. Congenital LCMV Virus: Mechanism of Brain Disease in a Rat Model of Congenital Viral Infection; University of Iowa: Iowa City, IA, USA, 2010. [Google Scholar]
- Bhattacharya, A.; Hegazy, A.N.; Deigendesch, N.; Kosack, L.; Cupovic, J.; Kandasamy, R.K.; Hildebrandt, A.; Merkler, D.; Kühl, A.A.; Vilagos, B. Superoxide dismutase 1 protects hepatocytes from type I interferon-driven oxidative damage. Immunity 2015, 43, 974–986. [Google Scholar] [CrossRef]
- Jamieson, D.J.; Kourtis, A.P.; Bell, M.; Rasmussen, S.A. Lymphocytic choriomeningitis virus: An emerging obstetric pathogen? Am. J. Obstet. Gynecol. 2006, 194, 1532–1536. [Google Scholar] [CrossRef]
- Mathews, S. S.; Albert, R.R.; Job, A. Audio-vestibular function in human immunodeficiency virus infected patients in India. Indian J. Sex. Transm. Dis. Aids 2012, 33, 98. [Google Scholar] [CrossRef]
- Chandrasekhar, S.S.; Connelly, P.E.; Brahmbhatt, S.S.; Shah, C.S.; Kloser, P.C.; Baredes, S. Otologic and audiologic evaluation of human immunodeficiency virus-infected patients. Am. J. Otolaryngol. 2000, 21, 1–9. [Google Scholar] [CrossRef]
- Van der Westhuizen, Y.; Swanepoel, D.W.; Heinze, B.; Hofmeyr, L.M. Auditory and otological manifestations in adults with HIV/AIDS. Int. J. Audiol. 2013, 52, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hahn, Y.K.; Podhaizer, E.M.; Hauser, K.F.; Knapp, P.E. HIV-1 alters neural and glial progenitor cell dynamics in the central nervous system: Coordinated response to opiates during maturation. Glia 2012, 60, 1871–1887. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Huertas, M.R.; Callejas, S.; Abia, D.; Mateos, E.; Dopazo, A.; Alcami, J.; Coiras, M. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucleic Acids Res. 2010, 38, 3287–3307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Martin, G.V.; Kelley, M.W.; Gridley, T. A mutation in the Lunatic fringe gene suppresses the effects of a Jagged2 mutation on inner hair cell development in the cochlea. Curr. Biol. 2000, 10, 659–662. [Google Scholar] [CrossRef]
- Bradley, T.; Ferrari, G.; Haynes, B.F.; Margolis, D.M.; Browne, E.P. Single-cell analysis of quiescent HIV infection reveals host transcriptional profiles that regulate proviral latency. Cell Rep. 2018, 25, 107–117. [Google Scholar] [CrossRef]
- Marra, C.M.; Wechkin, H.A.; Longstreth, W.; Rees, T.S.; Syapin, C.L.; Gates, G.A. Hearing loss and antiretroviral therapy in patients infected with HIV-1. Arch. Neurol. 1997, 54, 407–410. [Google Scholar] [CrossRef]
- Vincenti, V.; Pasanisi, E.; Bacciu, A.; Giordano, D.; Di Lella, F.; Guida, M.; Bacciu, S. Cochlear Implantation in a Human Immunodeficiency Virus-Infected Patient. Laryngoscope 2005, 115, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Dunmade, A.; Segun-Busari, S.; Olajide, T.; Ologe, F. Profound bilateral sensorineural hearing loss in Nigerian children: Any shift in etiology? J. Deaf. Stud. Deaf. Educ. 2007, 12, 112–118. [Google Scholar] [CrossRef]
- McKenna, M.J. Measles, mumps, and sensorineural hearing loss. Ann. N. Y. Acad. Sci. 1997, 830, 291–298. [Google Scholar] [CrossRef]
- Stephenson, J. Will the current measles vaccines ever eradicate measles? Expert Rev. Vaccines 2002, 1, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Karosi, T.; Kónya, J.; Petkó, M.; Sziklai, I. Histologic otosclerosis is associated with the presence of measles virus in the stapes footplate. Otol. Neurotol. 2005, 26, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, M.J.; Parmigiani, G.; Griffin, D.E. Gene expression patterns in dendritic cells infected with measles virus compared with other pathogens. Proc. Natl. Acad. Sci. USA 2006, 103, 3363–3368. [Google Scholar] [CrossRef]
- Kanra, G.; Kara, A.; Cengiz, A.B.; Isk, P.; Ceyhan, M.; Atas, A. Mumps meningoencephalitis effect on hearing. Pediatric Infect. Dis. J. 2002, 21, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Best, J.; MacMahon, E. Mumps and the UK epidemic 2005. BMJ 2005, 330, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ogawa, H.; Baba, Y.; Suzuki, T.; Yamada, N.; Omori, K. Cochlear implantation in a case of bilateral sensorineural hearing loss due to mumps. Fukushima J. Med Sci. 2009, 55, 32–38. [Google Scholar] [CrossRef][Green Version]
- Hayes, E.B.; Komar, N.; Nasci, R.S.; Montgomery, S.P.; O’Leary, D.R.; Campbell, G. L. Epidemiology and transmission dynamics of West Nile virus disease. Emerg. Infect. Dis. 2005, 11, 1167. [Google Scholar] [CrossRef]
- Jamison, S.C.; Michaels, S.R.; Ratard, R.; Sweet, J.M.; deBoisblanc, B.P. A 41-year-old HIV-positive man with acute onset of quadriplegia after West Nile virus infection. South. Med J. 2007, 100, 1051–1053. [Google Scholar] [CrossRef]
- McBride, W.; Gill, K.R.; Wiviott, L. West Nile Virus infection with hearing loss. J. Infect. 2006, 53, e203–e205. [Google Scholar] [CrossRef]
- Bourgeois, M. A.; Denslow, N.D.; Seino, K.S.; Barber, D.S.; Long, M.T. Gene expression analysis in the thalamus and cerebrum of horses experimentally infected with West Nile virus. PLoS ONE 2011, 6, e24371. [Google Scholar] [CrossRef]
- Aleksic, S.; Budzilovich, G.; Lieberman, A. Herpes zoster oticus and facial paralysis (Ramsay Hunt syndrome): Clinico-pathologic study and review of literature. J. Neurol. Sci. 1973, 20, 149–159. [Google Scholar] [CrossRef]
- Arnold, N.; Girke, T.; Sureshchandra, S.; Messaoudi, I. Acute simian varicella virus infection causes robust and sustained changes in gene expression in the sensory ganglia. J. Virol. 2016, 90, 10823–10843. [Google Scholar] [CrossRef] [PubMed]
- Nielsen-Saines, K.; Brasil, P.; Kerin, T.; Vasconcelos, Z.; Gabaglia, C.R.; Damasceno, L.; Pone, M.; de Carvalho, L.M.A.; Pone, S.M.; Zin, A.A. Delayed childhood neurodevelopment and neurosensory alterations in the second year of life in a prospective cohort of ZIKV-exposed children. Nat. Med. 2019, 25, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Glover, K.K.; Zahedi-Amiri, A.; Lao, Y.; Spicer, V.; Klonisch, T.; Coombs, K.M. Zika Infection Disrupts Proteins Involved in the Neurosensory System. Front. Cell Dev. Biol. 2020, 8, 571. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, X.-T.; Wang, G.; Cheng, X.; Yan, Y.; Fu, Y.-J.; Yang, X.; Jiang, Z. Zika virus induces abnormal cranial osteogenesis by negatively affecting cranial neural crest development. Infect. Genet. Evol. 2019, 69, 176–189. [Google Scholar] [CrossRef]
- Devhare, P.; Meyer, K.; Steele, R.; Ray, R.B.; Ray, R. Zika virus infection dysregulates human neural stem cell growth and inhibits differentiation into neuroprogenitor cells. Cell Death Dis. 2017, 8, e3106. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Javed, A.O.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef]
- Thawani, A.; Sirohi, D.; Kuhn, R.J.; Fekete, D.M. Zika virus can strongly infect and disrupt secondary organizers in the ventricular zone of the embryonic chicken brain. Cell Rep. 2018, 23, 692–700. [Google Scholar] [CrossRef]
- Zahedi-Amiri, A.; Sequiera, G.L.; Dhingra, S.; Coombs, K.M. Influenza a virus-triggered autophagy decreases the pluripotency of human-induced pluripotent stem cells. Cell Death Dis. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Terrier, O.; Textoris, J.; Carron, C.; Marcel, V.; Bourdon, J.-C.; Rosa-Calatrava, M. Host microRNA molecular signatures associated with human H1N1 and H3N2 influenza A viruses reveal an unanticipated antiviral activity for miR-146a. J. Gen. Virol. 2013, 94, 985–995. [Google Scholar] [CrossRef]
- Kroeker, A.L.; Ezzati, P.; Halayko, A.J.; Coombs, K.M. Response of primary human airway epithelial cells to influenza infection: A quantitative proteomic study. J. Proteome Res. 2012, 11, 4132–4146. [Google Scholar] [CrossRef] [PubMed]
- Koumpa, F.S.; Forde, C.T.; Manjaly, J.G. Sudden irreversible hearing loss post COVID-19. BMJ Case Rep. 2020, 13, e238419. [Google Scholar] [CrossRef] [PubMed]
- Rhman, S.S.A.; Wahid, A.A.A. COVID-19 and sudden sensorineural hearing loss, a case report. Otolaryngol. Case Rep. 2020, 16, 100198. [Google Scholar] [CrossRef]
- Degen, C.; Lenarz, T.; Willenborg, K. Acute Profound Sensorineural Hearing Loss after COVID-19 Pneumonia; Mayo Clinic Proceedings, 2020; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1801–1803. [Google Scholar]
- Hachim, I.Y.; Hachim, M.Y.; Talaat, I.M.; López-Ozuna, V.M.; Sharif-Askari, N.S.; Halwani, R.; Hamid, Q. The molecular basis of gender variations in mortality rates associated with the novel coronavirus (COVID-19) outbreak. Preprints 2020. [Google Scholar] [CrossRef]
- Fagone, P.; Ciurleo, R.; Lombardo, S.D.; Iacobello, C.; Palermo, C.I.; Shoenfeld, Y.; Bendtzen, K.; Bramanti, P.; Nicoletti, F. Transcriptional landscape of SARS-CoV-2 infection dismantles pathogenic pathways activated by the virus, proposes unique sex-specific differences and predicts tailored therapeutic strategies. Autoimmun. Rev. 2020, 19, 102571. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karimi-Boroujeni, M.; Zahedi-Amiri, A.; Coombs, K.M. Embryonic Origins of Virus-Induced Hearing Loss: Overview of Molecular Etiology. Viruses 2021, 13, 71. https://doi.org/10.3390/v13010071
Karimi-Boroujeni M, Zahedi-Amiri A, Coombs KM. Embryonic Origins of Virus-Induced Hearing Loss: Overview of Molecular Etiology. Viruses. 2021; 13(1):71. https://doi.org/10.3390/v13010071
Chicago/Turabian StyleKarimi-Boroujeni, Maryam, Ali Zahedi-Amiri, and Kevin M. Coombs. 2021. "Embryonic Origins of Virus-Induced Hearing Loss: Overview of Molecular Etiology" Viruses 13, no. 1: 71. https://doi.org/10.3390/v13010071
APA StyleKarimi-Boroujeni, M., Zahedi-Amiri, A., & Coombs, K. M. (2021). Embryonic Origins of Virus-Induced Hearing Loss: Overview of Molecular Etiology. Viruses, 13(1), 71. https://doi.org/10.3390/v13010071