Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA Data
2.2. Patient Demographics
2.3. Sequence Data Quality Control and Trimming
2.4. Retrotranscriptome Quantification
2.5. Transcriptome Quantification
2.6. Determination of HPV Status
2.7. Gene Set Enrichment Analysis
2.8. Clustering and Survival Analysis
3. Results
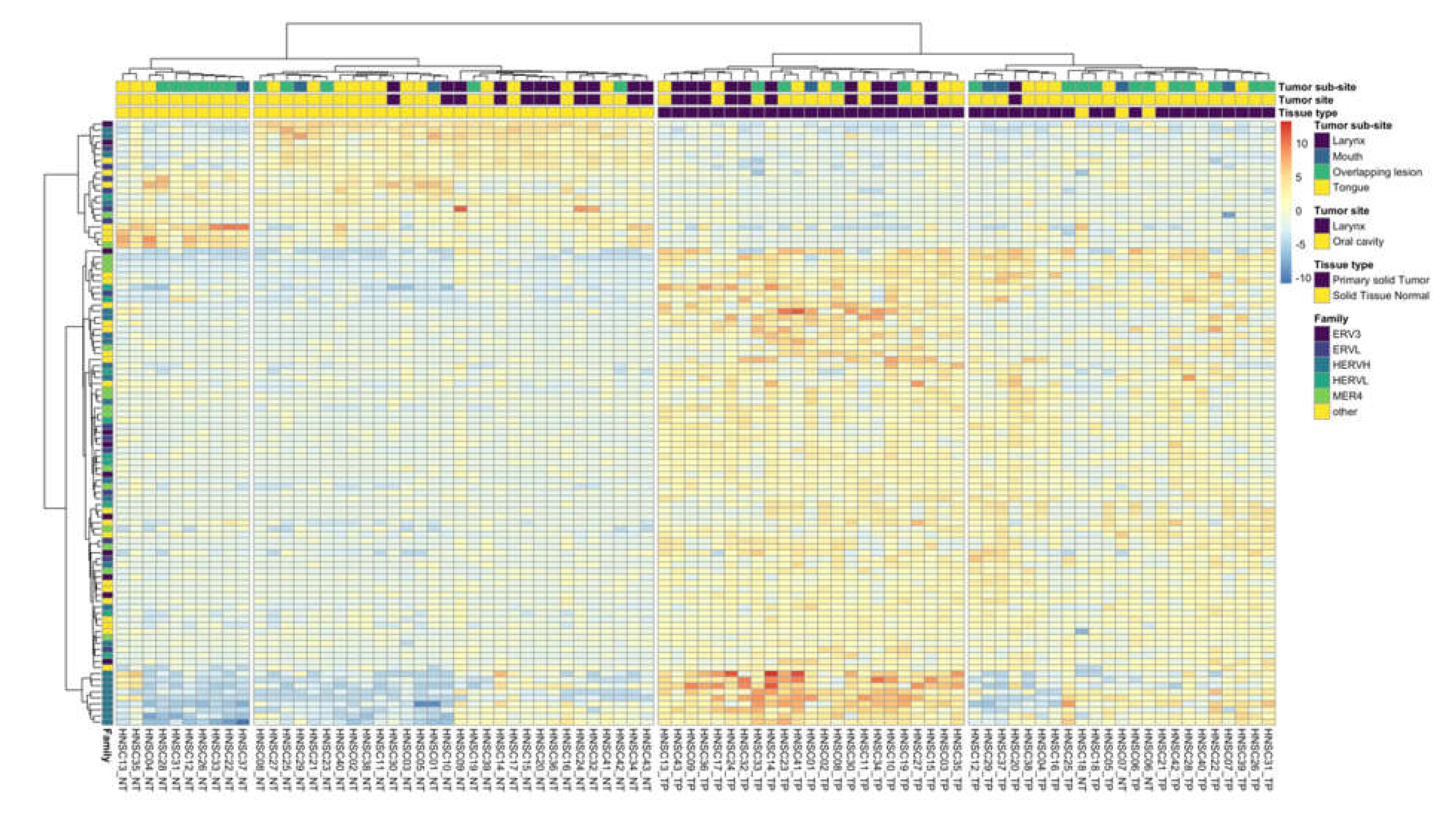
3.1. HERV Expression
3.2. HERV Expression, Phenotypic and Genotypic Associations
3.3. Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flockerzi, A.; Ruggieri, A.; Frank, O.; Sauter, M.; Maldener, E.; Kopper, B.; Wullich, B.; Seifarth, W.; Muller-Lantzsch, N.; Leib-Mosch, C.; et al. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics 2008, 9, 354. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montojo, M.; Doucet-O’Hare, T.; Henderson, L.; Nath, A. Human endogenous retrovirus-K (HML-2): A comprehensive review. Crit. Rev. Microbiol. 2018, 44, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J.; Rosenkrantz, J.L.; Carbone, L.; Chavez, S.L. Endogenous retroviruses: With us and against us. Front. Chem. 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A. Human endogenous retroviruses: Friend or foe? APMIS 2016, 124, 4–10. [Google Scholar] [CrossRef]
- Singh, S.; Kaye, S.; Gore, M.E.; McClure, M.O.; Bunker, C.B. The role of human endogenous retroviruses in melanoma. Br. J. Dermatol. 2009, 161, 1225–1231. [Google Scholar] [CrossRef]
- Büscher, K.; Trefzer, U.; Hofmann, M.; Sterry, W.; Kurth, R.; Denner, J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005, 65, 4172–4180. [Google Scholar] [CrossRef]
- Contreras-Galindo, R.; Kaplan, M.H.; Leissner, P.; Verjat, T.; Ferlenghi, I.; Bagnoli, F.; Giusti, F.; Dosik, M.H.; Hayes, D.F.; Gitlin, S.D.; et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J. Virol. 2008, 82, 9329–9336. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Frost, A.R.; Jian, B.; Epp, L.; Lu, D.W.; Johanning, G.L. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene 2003, 22, 1528–1535. [Google Scholar] [CrossRef]
- Downey, R.F.; Sullivan, F.J.; Wang-Johanning, F.; Ambs, S.; Giles, F.J.; Glynn, S.A. Human endogenous retrovirus K and cancer: Innocent bystander or tumorigenic accomplice? Int. J. Cancer 2015, 137, 1249–1257. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Liu, J.; Rycaj, K.; Huang, M.; Tsai, K.; Rosen, D.G.; Chen, D.-T.; Lu, D.W.; Barnhart, K.F.; Johanning, G.L. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int. J. Cancer 2007, 120, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liang, J.Q.; Zheng, S. Expressional activation and functional roles of human endogenous retroviruses in cancers. Rev. Med. Virol. 2019, 29, e2025. [Google Scholar] [CrossRef] [PubMed]
- Bannert, N.; Hofmann, H.; Block, A.; Hohn, O. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front. Microbiol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Serafini, M.S.; Lopez-Perez, L.; Fico, G.; Licitra, L.; De Cecco, L.; Resteghini, C. Transcriptomics and Epigenomics in head and neck cancer: Available repositories and molecular signatures. Cancers Head Neck 2020, 5, 2. [Google Scholar] [CrossRef]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef]
- Panda, A.; de Cubas, A.A.; Stein, M.; Riedlinger, G.; Kra, J.; Mayer, T.; Smith, C.C.; Vincent, B.G.; Serody, J.S.; Beckermann, K.E.; et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Attig, J.; Young, G.R.; Hosie, L.; Perkins, D.; Encheva-Yokoya, V.; Stoye, J.P.; Snijders, A.P.; Ternette, N.; Kassiotis, G. LTR retroelement expansion of the human cancer transcriptome and immunopeptidome revealed by de novo transcript assembly. Genome Res. 2019, 29, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.-J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef] [PubMed]
- Bendall, M.L.; de Mulder, M.; Lecanda-Sánchez, A.; Pérez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Terán, G.; Crandall, K.A.; Ormsby, C.E.; et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Picard Tools - By Broad Institute - GitHub Pages. Available online: http://broadinstitute.github.io/picard/ (accessed on 28 August 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Soneson, C.; Robinson, M.D. Importing transcript abundance datasets with tximport. Dim Txi. Inf. Rep. Sample1 2017, 1, 5. [Google Scholar]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Sherrill-Mix, S. GitHub - sherrillmix/taxonomizr: Parse NCBI taxonomy and accessions to find taxonomix assignments. Available online: https://github.com/sherrillmix/taxonomizr (accessed on 27 August 2020).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Therneau, T.M. GitHub – therneau/survival: Survival package for R. Available online: https://github.com/therneau/survival (accessed on 27 August 2020).
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer Science & Business Media: New York, NY, USA, 2013. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 3698. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biecek, P. GitHub – kassambara/survminer: Survival Analysis and Visualization. Available online: https://github.com/kassambara/survminer/ (accessed on 27 August 2020).
- Kolde, R. pheatmap: Pretty Heatmaps. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 27 August 2020).
- Garnier, S.; Ross, N.; Rudis, B.; Sciaini, M.; Scherer, C. viridis: Default Color Maps from ’matplotlib’. Available online: https://cran.r-project.org/web/packages/viridis/ (accessed on 27 August 2020).
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes. Available online: https://cran.r-project.org/web/packages/RColorBrewer/index.html (accessed on 27 August 2020).
- Chuang, S.C.; Agudo, A.; Ahrens, W.; Anantharaman, D.; Benhamou, S.; Boccia, S.; Chen, C.; Conway, D.I.; Fabianova, E.; Hayes, R.B.; et al. Sequence Variants and the Risk of Head and Neck Cancer: Pooled Analysis in the Inhance Consortium. Front. Oncol. 2011, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.B.; Bhat, A.R.; James, B.L.; Govindan, S.V.; Mathew, R.; Ravindra, D.R.; Hedne, N.; Illiayaraja, J.; Kekatpure, V.; Khora, S.S.; et al. Meta-Analyses of Microarray Datasets Identifies ANO1 and FADD as Prognostic Markers of Head and Neck Cancer. PLoS ONE 2016, 11, e0147409. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Wang-Johanning, F.; Li, M.; Esteva, F.J.; Hess, K.R.; Yin, B.; Rycaj, K.; Plummer, J.B.; Garza, J.G.; Ambs, S.; Johanning, G.L. Human endogenous retrovirus type K antibodies and mRNA as serum biomarkers of early-stage breast cancer. Int. J. Cancer 2014, 134, 587–595. [Google Scholar] [CrossRef]
- Zhao, J.; Rycaj, K.; Geng, S.; Li, M.; Plummer, J.B.; Yin, B.; Liu, H.; Xu, X.; Zhang, Y.; Yan, Y.; et al. Expression of Human Endogenous Retrovirus Type K Envelope Protein is a Novel Candidate Prognostic Marker for Human Breast Cancer. Genes Cancer 2011, 2, 914–922. [Google Scholar] [CrossRef]
- Liang, Q.; Xu, Z.; Xu, R.; Wu, L.; Zheng, S. Expression patterns of non-coding spliced transcripts from human endogenous retrovirus HERV-H elements in colon cancer. PLoS ONE 2012, 7, e29950. [Google Scholar] [CrossRef]
- Yi, J.-M.; Kim, H.-M.; Kim, H.-S. Human endogenous retrovirus HERV-H family in human tissues and cancer cells: Expression, identification, and phylogeny. Cancer Lett. 2006, 231, 228–239. [Google Scholar] [CrossRef]
- Hu, S.; Yuan, H.; Li, Z.; Zhang, J.; Wu, J.; Chen, Y.; Shi, Q.; Ren, W.; Shao, N.; Ying, X. Transcriptional response profiles of paired tumor-normal samples offer novel perspectives in pan-cancer analysis. Oncotarget 2017, 8, 41334–41347. [Google Scholar] [CrossRef]
- Huang, X.; Stern, D.F.; Zhao, H. Transcriptional profiles from paired normal samples offer complementary information on cancer patient survival--evidence from TCGA pan-cancer data. Sci. Rep. 2016, 6, 20567. [Google Scholar] [CrossRef]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Ahn, J.B.; Rha, S.Y.; Chung, H.C.; Park, K.H.; Kim, T.S.; Kim, N.K.; Shin, S.J. High KLF4 level in normal tissue predicts poor survival in colorectal cancer patients. World J. Surg. Oncol. 2014, 12, 232. [Google Scholar] [CrossRef] [PubMed]
- Román-Pérez, E.; Casbas-Hernández, P.; Pirone, J.R.; Rein, J.; Carey, L.A.; Lubet, R.A.; Mani, S.A.; Amos, K.D.; Troester, M.A. Gene expression in extratumoral microenvironment predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012, 14, R51. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.; Ge, X.; de Las Morenas, A.; Tripathi, A.; Rosenberg, C.L. Gene expression profiles of estrogen receptor-positive and estrogen receptor-negative breast cancers are detectable in histologically normal breast epithelium. Clin. Cancer Res. 2011, 17, 236–246. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolbe, A.R.; Bendall, M.L.; Pearson, A.T.; Paul, D.; Nixon, D.F.; Pérez-Losada, M.; Crandall, K.A. Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival. Viruses 2020, 12, 956. https://doi.org/10.3390/v12090956
Kolbe AR, Bendall ML, Pearson AT, Paul D, Nixon DF, Pérez-Losada M, Crandall KA. Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival. Viruses. 2020; 12(9):956. https://doi.org/10.3390/v12090956
Chicago/Turabian StyleKolbe, Allison R., Matthew L. Bendall, Alexander T. Pearson, Doru Paul, Douglas F. Nixon, Marcos Pérez-Losada, and Keith A. Crandall. 2020. "Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival" Viruses 12, no. 9: 956. https://doi.org/10.3390/v12090956
APA StyleKolbe, A. R., Bendall, M. L., Pearson, A. T., Paul, D., Nixon, D. F., Pérez-Losada, M., & Crandall, K. A. (2020). Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival. Viruses, 12(9), 956. https://doi.org/10.3390/v12090956