Family Level Phylogenies Reveal Relationships of Plant Viruses within the Order Bunyavirales




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analysis of Bunyavirales
2.2. Analysis of the Untranslated Regions (UTRs) of RNA Segments
2.3. Pairwise Sequence Alignment and Identity Score Calculation
2.4. Co-Phylogenetic Analysis
3. Results
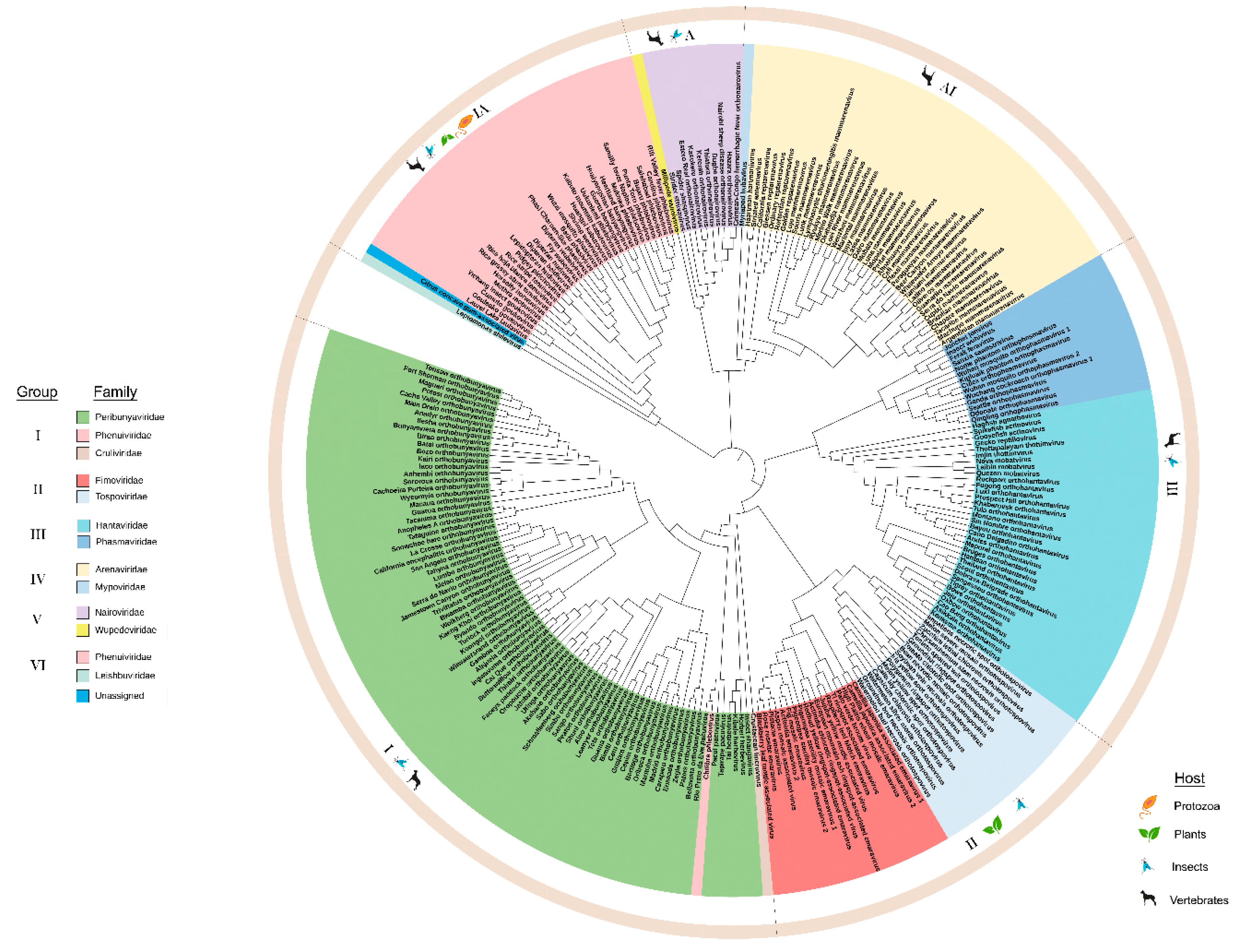
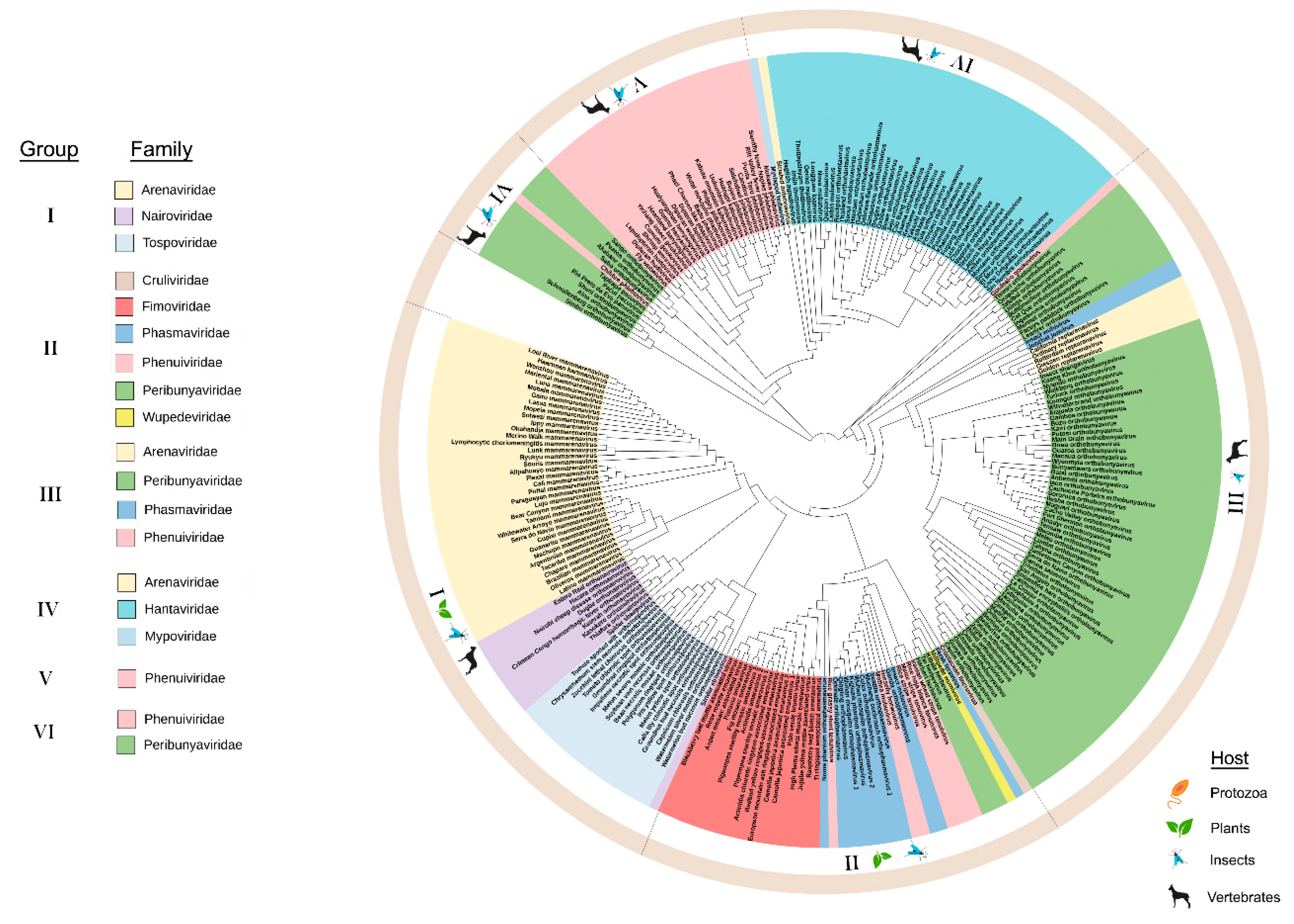
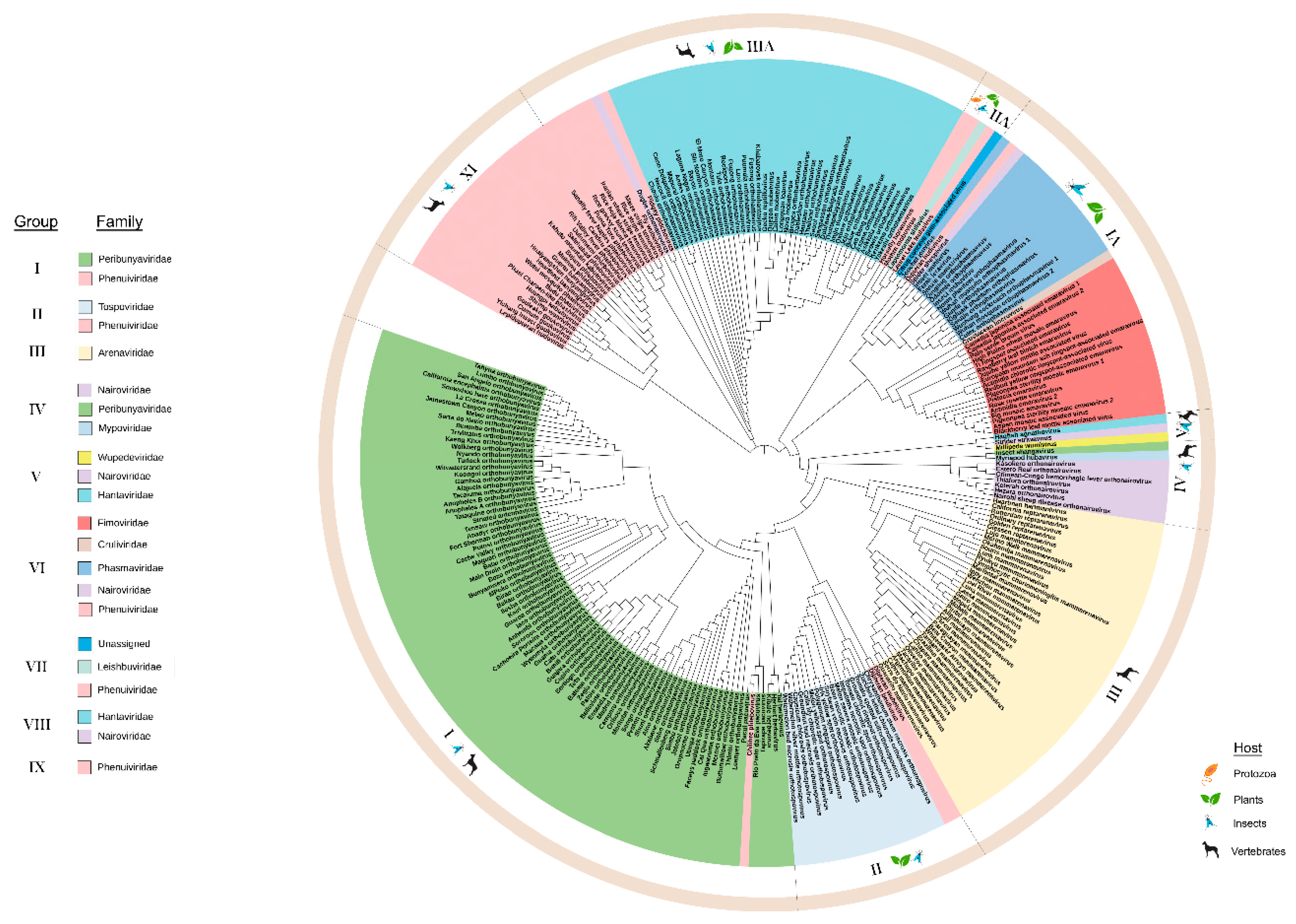
3.1. Phylogeny and Domain Analysis of RNA-Dependent RNA Polymerase (RdRp), Glycoprotein Precursor (preGP), Nucleocapsid Proteins (N), and Movement Proteins (MP) of Bunyavirales Members
3.1.1. Phylogeny of RdRp
3.1.2. Phylogeny of preGP
3.1.3. Phylogeny of Nucleocapsid (N) Proteins
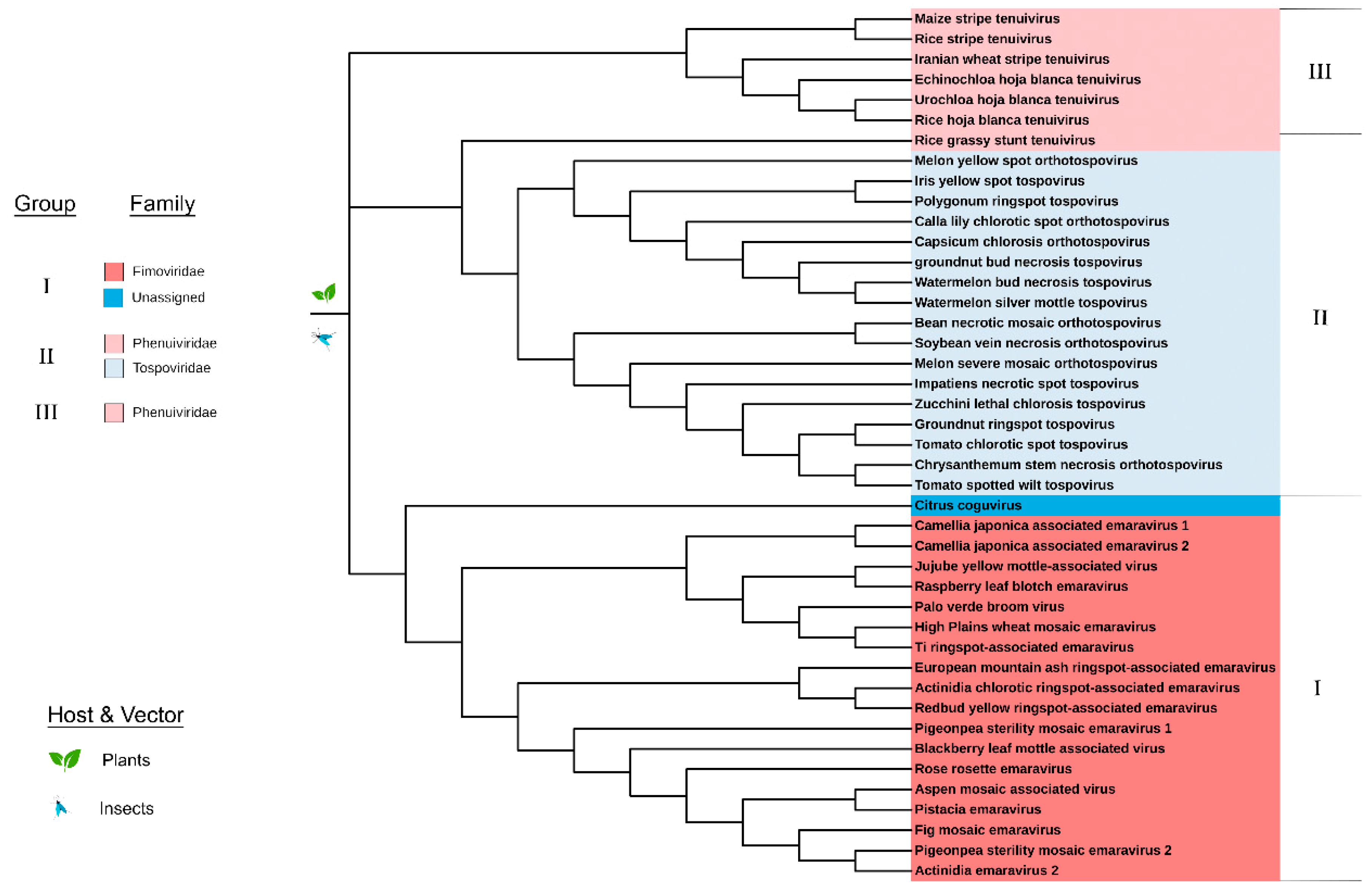
3.2. Phylogeny of MP of Plant Virus Genera Orthotospovirus, Emaravirus and Tenuivirus
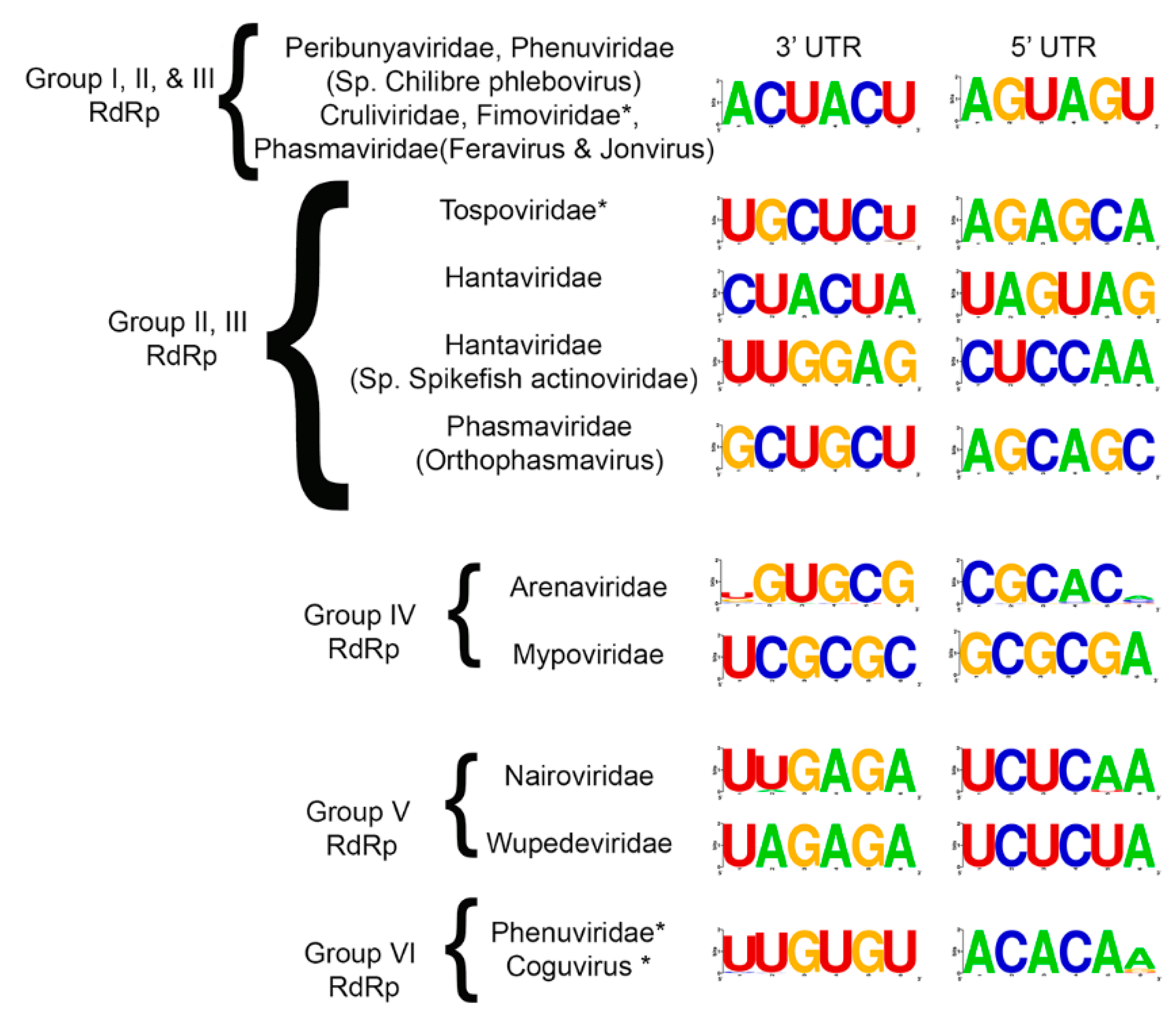
3.3. Common Features of Complementary 3′ and 5′ Terminal Regions of Genome Segments
3.4. Cophylogenetic Analysis and Host Range Evolution
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wichgers Schreur, P.J.; Kormelink, R.; Kortekaas, J. Genome packaging of the Bunyavirales. Curr. Opin. Virol. 2018, 33, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Minutolo, M.; De Stradis, A.; Palmisano, F.; Alioto, D.; Di Serio, F. The first phlebo-like virus infecting plants: A case study on the adaptation of negative-stranded RNA viruses to new hosts. Mol. Plant Pathol. 2018, 19, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Zicca, S.; Minutolo, M.; Saponari, M.; Alioto, D.; Di Serio, F. A negative-stranded RNA virus infecting citrus trees: The second member of a new genus within the order bunyavirales. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 2015, 1–26. [Google Scholar] [CrossRef]
- Guterres, A.; de Oliveira, R.C.; Fernandes, J.; de Lemos, E.R.S.; Schrago, C.G. New bunya-like viruses: Highlighting their relations. Infect. Genet. Evol. 2017, 49, 164–173. [Google Scholar] [CrossRef]
- Ballinger, M.J.; Taylor, D.J. Evolutionary persistence of insect bunyavirus infection despite host acquisition and expression of the viral nucleoprotein gene. Virus Evol. 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Whitfield, A.E.; Huot, O.B.; Martin, K.M.; Kondo, H.; Dietzgen, R.G. Plant rhabdoviruses—Their origins and vector interactions. Curr. Opin. Virol. 2018, 33, 198–207. [Google Scholar] [CrossRef]
- Bojko, J.; Subramaniam, K.; Waltzek, T.B.; Stentiford, G.D.; Behringer, D.C. Genomic and developmental characterisation of a novel bunyavirus infecting the crustacean Carcinus maenas. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Käfer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M.; et al. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS Pathog. 2019, 15, e1008224. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V. Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements. Microbiol. Mol. Biol. Rev. 2014, 78, 278–303. [Google Scholar] [CrossRef] [PubMed]
- Kormelink, R.; Garcia, M.L.; Goodin, M.; Sasaya, T.; Haenni, A.L. Negative-strand RNA viruses: The plant-infecting counterparts. Virus Res. 2011, 162, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benk, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and evolution of the global RNA virome. mBio 2018, 9, 1–31. [Google Scholar] [CrossRef]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, 22–28. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- MAFFT Version 7. Available online: https://mafft.cbrc.jp/alignment/server/ (accessed on 12 August 2020).
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools—An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011. [Google Scholar] [CrossRef]
- Guindon, S.; Delsuc, F.; Dufayard, J.-F.; Gascuel, O. Estimating Maximum Likelihood Phylogenies with PhyML. In Bioinformatics for DNA Sequence Analysis; Posada, D., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 113–137. ISBN 978-1-59745-251-9. [Google Scholar]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A multiplatform graphical user Interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- iTOL. Available online: https://itol.embl.de/ (accessed on 9 August 2020).
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- WebLogo 3. Available online: http://weblogo.threeplusone.com/ (accessed on 27 April 2020).
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Conow, C.; Fielder, D.; Ovadia, Y.; Libeskind-Hadas, R. Jane: A new tool for the cophylogeny reconstruction problem. Algorithms Mol. Biol. 2010, 5, 1–10. [Google Scholar] [CrossRef]
- Taxonomy. Available online: https://www.ncbi.nlm.nih.gov/taxonomy (accessed on 27 July 2020).
- Virus-Host Database. Available online: https://www.genome.jp/virushostdb/ (accessed on 27 July 2020).
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking virus genomes with host taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; Stevens, P.F.; et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016. [Google Scholar] [CrossRef]
- ANGIOSPERM PHYLOGENY WEBSITE, Version 14. Available online: http://www.mobot.org/MOBOT/research/APweb/ (accessed on 11 August 2020).
- Mielke-Ehret, N.; Mühlbach, H.-P. Emaravirus: A novel genus of multipartite, negative strand RNA plant viruses. Viruses 2012, 4, 1515–1536. [Google Scholar] [CrossRef]
- German, T.L.; Lorenzen, M.D.; Grubbs, N.; Whitfield, A.E. New technologies for studying negative-strand RNA viruses in plant and arthropod hosts. Mol. Plant Microbe Interact. 2020, 33, 382–393. [Google Scholar] [CrossRef]
- Dolja, V.V.; Koonin, E.V. Common origins and host-dependent diversity of plant and animal viromes. Curr. Opin. Virol. 2011, 1, 322–331. [Google Scholar] [CrossRef]
- Amroun, A.; Priet, S.; de Lamballerie, X.; Quérat, G. Bunyaviridae RdRps: Structure, motifs, and RNA synthesis machinery. Crit. Rev. Microbiol. 2017, 43, 753–778. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Weber, F.; de la Torre, J.C.; Reguera, J. Transcription and replication mechanisms of Bunyaviridae and Arenaviridae L proteins. Virus Res. 2017, 234, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, J.; Gao, G.F.; Tien, P.; Liu, W. Bunyavirales ribonucleoproteins: The viral replication and transcription machinery. Crit. Rev. Microbiol. 2018, 44, 522–540. [Google Scholar] [CrossRef]
- Guardado-Calvo, P.; Rey, F.A. The Envelope Proteins of the Bunyavirales. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2017; Volume 98, pp. 83–118. [Google Scholar]
- Brown, J.K.; Fauquet, C.M.; Briddon, R.W.; Zerbini, M.; Moriones, E.; Navas-Castillo, J. Bunyaviridae. In Virus Taxonomy; Elsevier: New York, NY, USA, 2012; pp. 725–741. [Google Scholar]
- Ishikawa, K.; Maejima, K.; Komatsu, K.; Netsu, O.; Keima, T.; Shiraishi, T.; Okano, Y.; Hashimoto, M.; Yamaji, Y.; Namba, S. Fig mosaic emaravirus p4 protein is involved in cell-to-cell movement. J. Gen. Virol. 2013, 94, 682–686. [Google Scholar] [CrossRef]
- Yu, C.; Karlin, D.G.; Lu, Y.; Wright, K.; Chen, J.; MacFarlane, S. Experimental and bioinformatic evidence that raspberry leaf blotch emaravirus P4 is a movement protein of the 30K superfamily. J. Gen. Virol. 2013, 94, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Cao, M.; Liu, W.; Ren, Y.; Zhou, X.; Wang, X. Two negative-strand RNA viruses identified in watermelon represent a novel clade in the order Bunyavirales. Front. Microbiol. 2017, 8, 1514. [Google Scholar] [CrossRef]
- Zhao, W.; Jiang, L.; Feng, Z.; Chen, X.; Huang, Y.; Xue, F.; Huang, C.; Liu, Y.; Li, F.; Liu, Y.; et al. Plasmodesmata targeting and intercellular trafficking of Tomato spotted wilt tospovirus movement protein NSm is independent of its function in HR induction. J. Gen. Virol. 2016, 97, 1990–1997. [Google Scholar] [CrossRef]
- Melcher, U. The “30K” superfamily of viral movement proteins. J. Gen. Virol. 2000, 81, 257–266. [Google Scholar] [CrossRef]
- Mushegian, A.R.; Elena, S.F. Evolution of plant virus movement proteins from the 30K superfamily and of their homologs integrated in plant genomes. Virology 2015, 476, 304–315. [Google Scholar] [CrossRef]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef]
- Terret-Welter, Z.; Bonnet, G.; Moury, B.; Gallois, J.-L. Analysis of tomato spotted wilt virus RNA-dependent RNA polymerase adaptative evolution and constrained domains using homology protein structure modelling. J. Gen. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Klemm, C.; Reguera, J.; Cusack, S.; Zielecki, F.; Kochs, G.; Weber, F. Systems to establish Bunyavirus genome replication in the absence of transcription. J. Virol. 2013, 87, 8205–8212. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, B.; Ding, Z.; Li, G.; Liu, M.; Zhu, D.; Sun, Y.; Dong, S.; Lou, Z. Distinct mechanism for the formation of the ribonucleoprotein complex of Tomato spotted wilt virus. J. Virol. 2017, 91, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kobayashi, Y. Evolution of complementary nucleotides in 5’ and 3’ untranslated regions of influenza A virus genomic segments. Infect. Genet. Evol. 2013, 13, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Hughes, H.R.; Russell, B.J.; Lambert, A.J. Genetic characterization of frijoles and chilibre species complex viruses (genus phlebovirus; Family phenuiviridae) and three unclassified new world phleboviruses. Am. J. Trop. Med. Hyg. 2020, 102, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Calisher, C.H.; Higgs, S. Viruses of the family Bunyaviridae: Are all available isolates reassortants? Virology 2013, 446, 207–216. [Google Scholar] [CrossRef]
- Wang, J.; Firth, C.; Amos-Ritchie, R.; Davis, S.S.; Yin, H.; Holmes, E.C.; Blasdell, K.R.; Walker, P.J. Evolutionary history of Simbu serogroup orthobunyaviruses in the Australian episystem. Virology 2019, 535, 32–44. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Ali, A. Mechanisms of plant virus evolution and identification of genetic bottlenecks: Impact on disease management. In Biotechnology and Plant Disease Management; CABI: Wallingford, UK, 2007; pp. 109–124. ISBN 9781845932886. [Google Scholar]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Holmes, E.C.; Zhang, Y.Z. The evolution and emergence of hantaviruses. Curr. Opin. Virol. 2015, 10, 27–33. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V. Expanding networks of RNA virus evolution. BMC Biol. 2012, 10, 54. [Google Scholar] [CrossRef]
- Liu, H.; Fu, Y.; Jiang, D.; Li, G.; Xie, J.; Cheng, J.; Peng, Y.; Ghabrial, S.A.; Yi, X. Widespread Horizontal Gene Transfer from Double-Stranded RNA Viruses to Eukaryotic Nuclear Genomes. J. Virol. 2010, 84, 11876–11887. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.G.; Reitz, S.R.; Perry, K.L.; Adkins, S. A natural M RNA reassortant arising from two species of plant- and insect-infecting bunyaviruses and comparison of its sequence and biological properties to parental species. Virology 2011, 413, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Senior, A.M.; Di Giallonardo, F.; Holmes, E.C. Virological factors that increase the transmissibility of emerging human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Duchêne, S.; Holmes, E.C. Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog. 2017, 13, e1006215. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Viruses at the Edge of Adaptation. Virology 2000, 270, 251–253. [Google Scholar] [CrossRef]
- McGavin, W.J.; Mitchell, C.; Cock, P.J.A.; Wright, K.M.; MacFarlane, S.A. Raspberry leaf blotch virus, a putative new member of the genus Emaravirus, encodes a novel genomic RNA. J. Gen. Virol. 2012, 93. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, S.; Han, T.; Fu, J.; Di Serio, F.; Cao, M. Identification and characterization of a novel emaravirus associated with jujube (Ziziphus jujuba Mill.) yellow mottle disease. Front. Microbiol. 2019, 10, 1–12. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herath, V.; Romay, G.; Urrutia, C.D.; Verchot, J. Family Level Phylogenies Reveal Relationships of Plant Viruses within the Order Bunyavirales. Viruses 2020, 12, 1010. https://doi.org/10.3390/v12091010
Herath V, Romay G, Urrutia CD, Verchot J. Family Level Phylogenies Reveal Relationships of Plant Viruses within the Order Bunyavirales. Viruses. 2020; 12(9):1010. https://doi.org/10.3390/v12091010
Chicago/Turabian StyleHerath, Venura, Gustavo Romay, Cesar D. Urrutia, and Jeanmarie Verchot. 2020. "Family Level Phylogenies Reveal Relationships of Plant Viruses within the Order Bunyavirales" Viruses 12, no. 9: 1010. https://doi.org/10.3390/v12091010
APA StyleHerath, V., Romay, G., Urrutia, C. D., & Verchot, J. (2020). Family Level Phylogenies Reveal Relationships of Plant Viruses within the Order Bunyavirales. Viruses, 12(9), 1010. https://doi.org/10.3390/v12091010