Genome Analysis of Two Novel Synechococcus Phages That Lack Common Auxiliary Metabolic Genes: Possible Reasons and Ecological Insights by Comparative Analysis of Cyanomyoviruses

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cyanophage Isolation
2.2. Phage Purification
2.3. Host Range
2.4. Morphological Study by Transmission Electron Microscopy
2.5. Genome Sequencing and Assembly
2.6. Genome Annotation and Analysis
2.7. Comparative Analysis of Cyanophage Genomes
3. Results and Discussion
3.1. Host Range and Phage Morphology
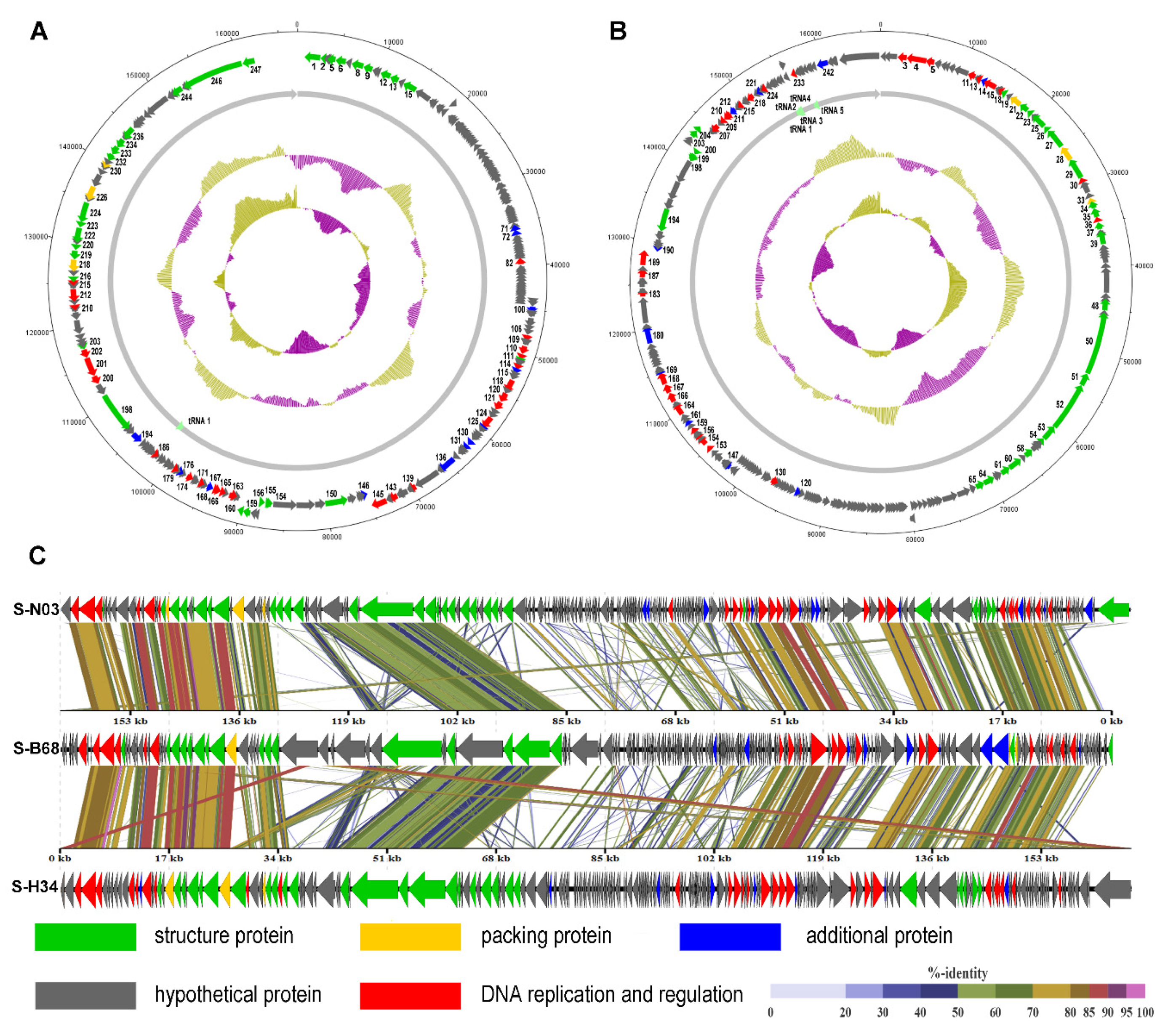
3.2. General Genomic Features
3.3. tRNA Genes
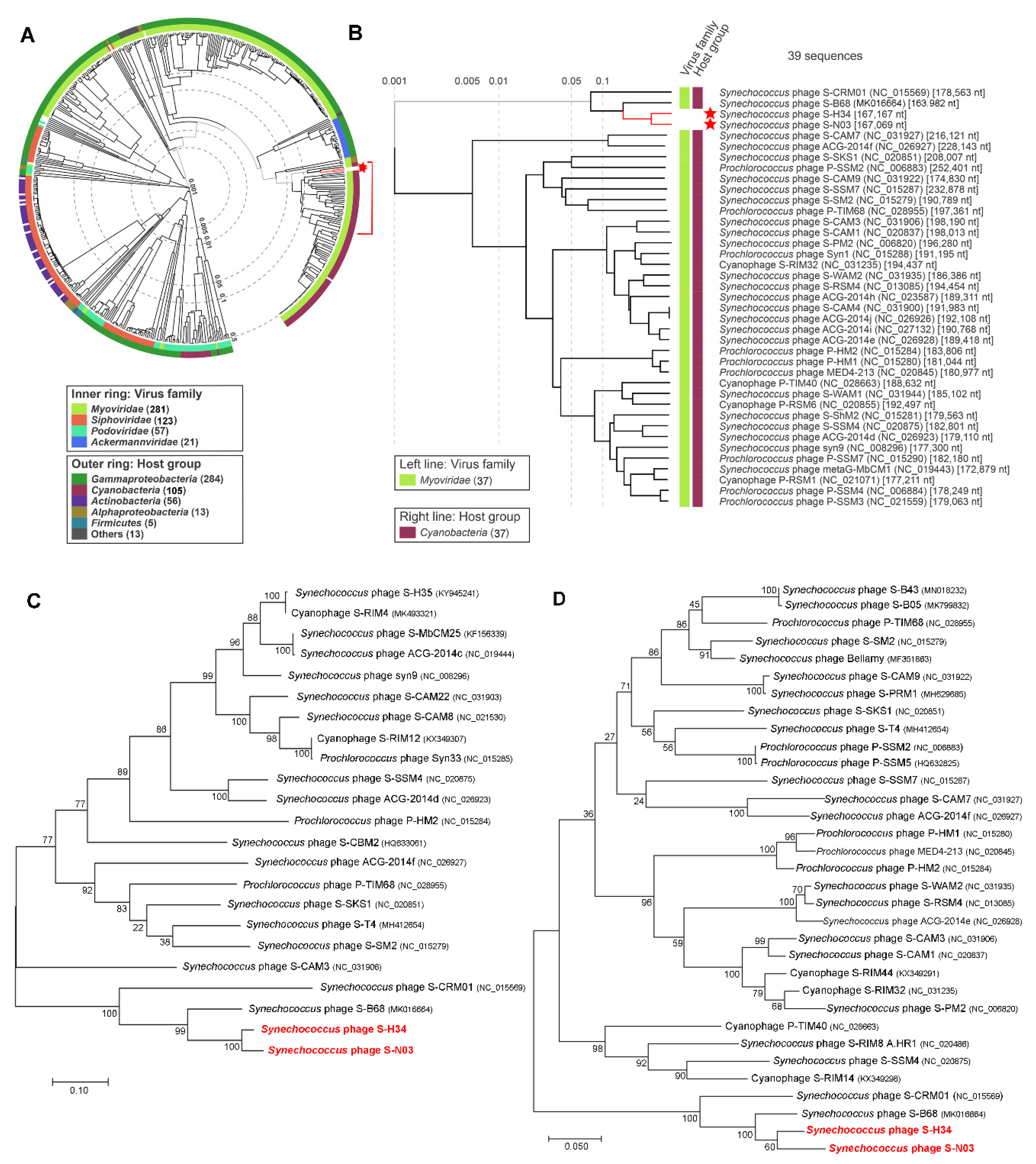
3.4. Genome Comparison and Phylogenetic Analysis
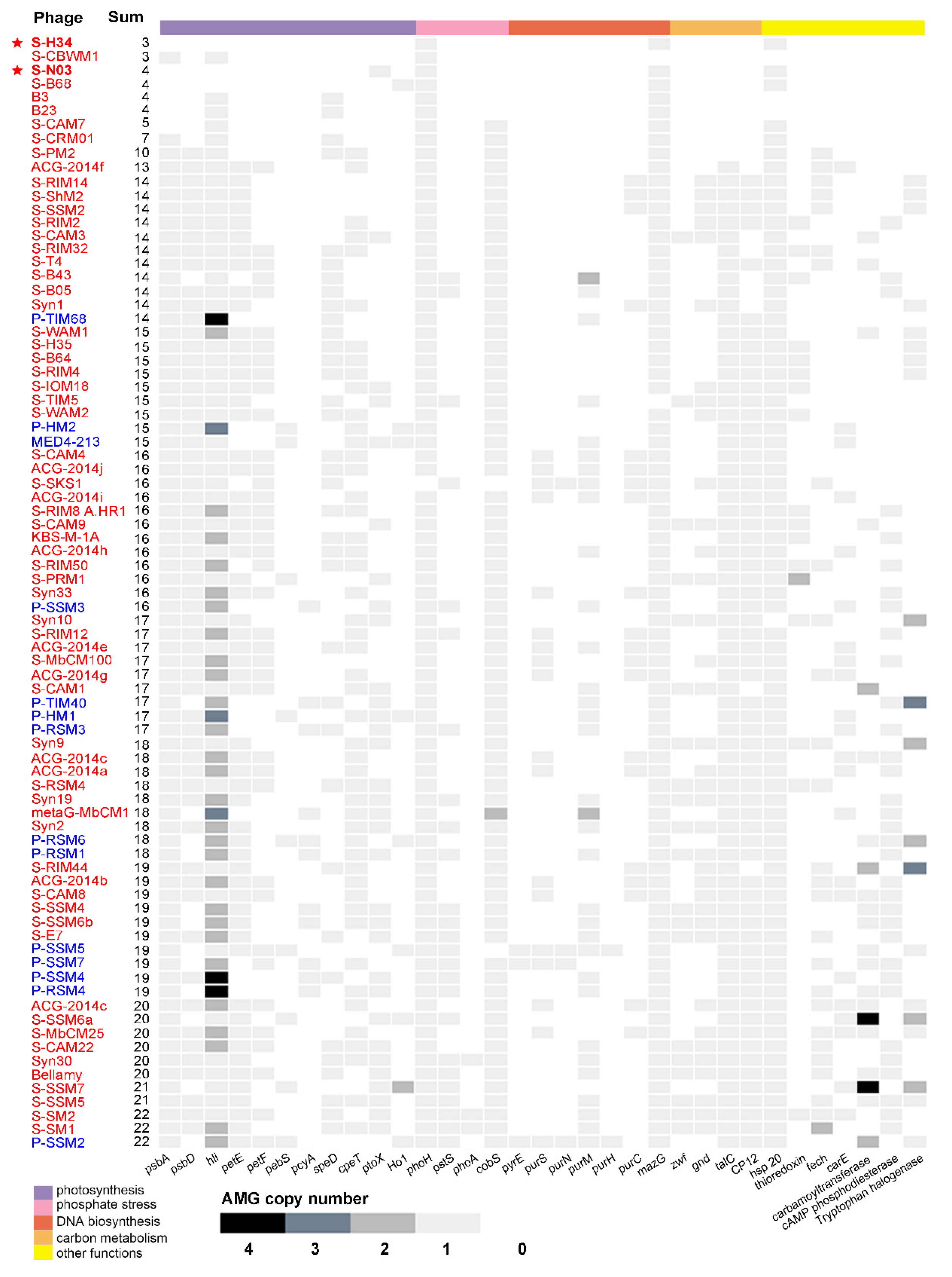
3.5. Auxiliary Metabolic Genes (AMGs)
3.5.1. MazG Gene (Pyrophosphatase)
3.5.2. phoH Gene (P-Starvation Inducible Protein)
3.5.3. Hsp Gene (Heat Shock Protein)
3.5.4. ptoX Gene (Plastoquinol Terminal Oxidase)
3.5.5. Lack of Photosynthetic AMGs
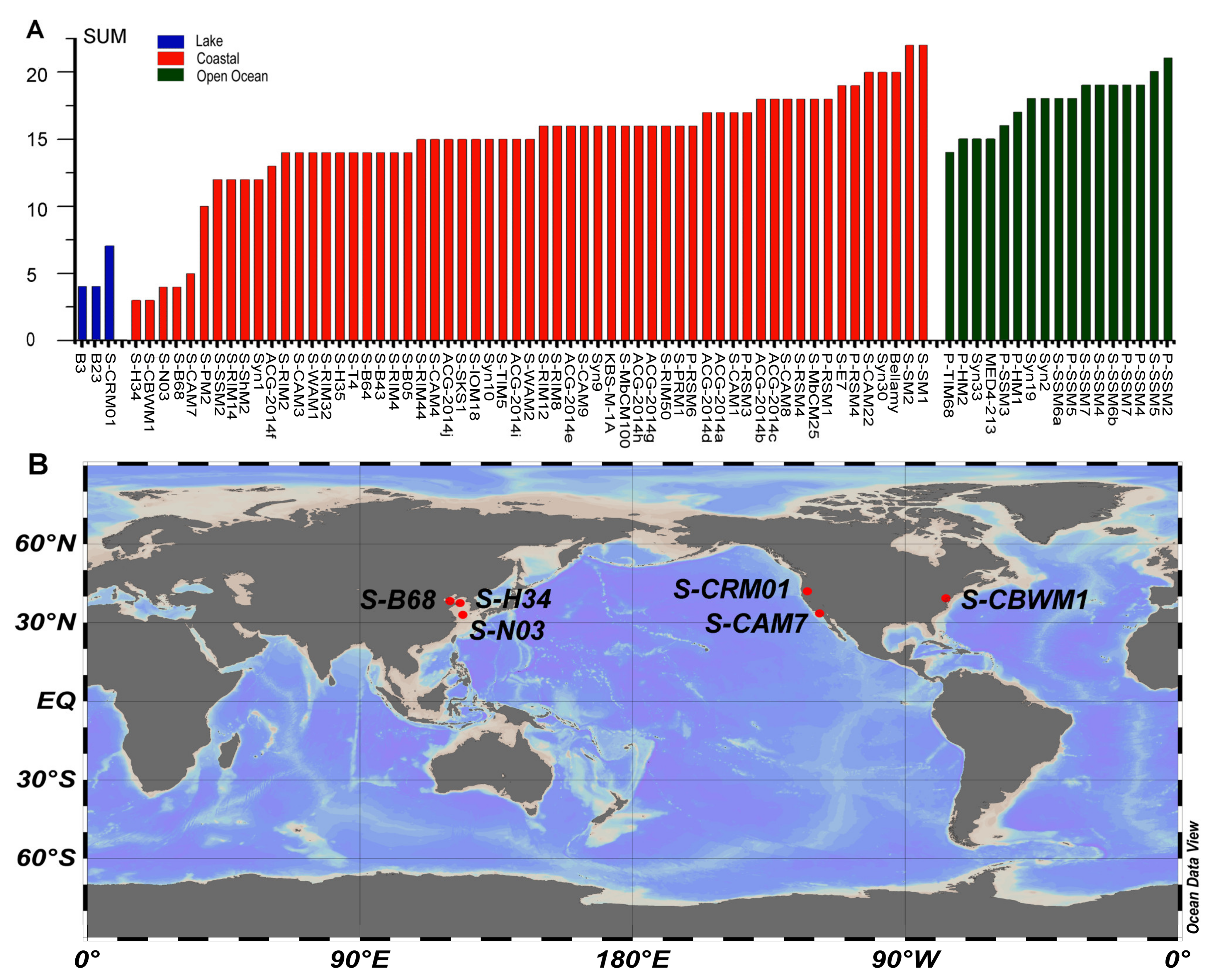
3.5.6. Low AMG Contents in S-H34 and S-N03
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Partensky, F.; Blanchot, J.; Vaulot, D. Differential distribution and ecology of Prochlorococcus and Synechococcus in oceanic waters: A review. Bull. Inst. Oceanogr. Monaco Numero Spec. 1999, 457–476. [Google Scholar]
- Scanlan, D.J. Marine picocyanobacteria. In Ecology of Cyanobacteria II; Whitton, B.A., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 503–533. [Google Scholar] [CrossRef]
- Moisander, P.H.; Breinart, R.A.; Hewson, I.; White, A.S.; Johnson, K.S.; Carolson, C.A.; Montoya, J.P.; Zehr, J.P. Unicellular cyanobacterial distributions broaden the oceanic N2 fixation domain. Science 2010, 327, 1512–1514. [Google Scholar] [CrossRef]
- Zehr, J.P.; Waterbury, J.B.; Turner, P.J.; Montoya, J.P.; Omoregie, E.; Steward, G.F.; Hansen, A.; Karl, D.M. Unicellular cyanobacteria fix N2 in the subtropical North Pacific Ocean. Nature 2001, 412, 635–638. [Google Scholar] [CrossRef]
- Lu, J.; Chen, F.; Hodson, R.E. Distribution, isolation, host specificity, and diversity of cyanophages infecting marine Synechococcus spp. in river estuaries. Appl. Environ. Microbiol. 2001, 67, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A.; Chan, A.M. Dynamics and distribution of cyanophages and their effect on marine Synechococcus spp. Appl. Environ. Microbiol. 1994, 60, 3167–3174. [Google Scholar] [CrossRef] [PubMed]
- Proctor, L.M.; Fuhrman, J.A. Viral mortality of marine bacteria and cyanobacteria. Nature 1990, 343, 60–62. [Google Scholar] [CrossRef]
- Suttle, C.A. The significance of viruses to mortality in aquatic microbial communities. Microb. Ecol. 1994, 28, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Hagay, E.; Mandel-Gutfreund, Y.; Béjà, O. Comparative metagenomics analyses reveal viral-induced shifts of host metabolism towards nucleotide biosysnthesis. Microbiome 2014, 2, 9. [Google Scholar] [CrossRef]
- Crummett, L.T.; Puxty, R.J.; Weihe, C. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 2016, 499, 219–229. [Google Scholar] [CrossRef]
- Lindell, D.; Jaffe, J.D.; Johnson, Z.I.; Church, G.M.; Chisholm, S.W. Photosynthesis genes in marine viruses yield proteins during host infection. Nature 2005, 438, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Chénard, C.; Suttle, C.A. Phylogenetic diversity of sequences of cyanophage photosynthetic gene psbA in marine and freshwaters. Appl. Environ. Microbiol. 2008, 74, 5317–5324. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.; Cook, A.; Millard, A.; Bailey, S.; Clokie, M. Marine ecosystems: Bacterial photosynthesis genes in a virus. Nature 2003, 424, 741. [Google Scholar] [CrossRef] [PubMed]
- Philosof, A.; Battchikova, N.; Aro, E.; Beja, O. Marine cyanophages: Tinkering with the electron transport chain. ISME J. 2011, 5, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.; Zeng, Q.; Kelly, L.; Huang, K.; Singer, A.; Stubbe, J.; Chisholm, S. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Nat. Acad. Sci. USA 2011, 108, E757–E764. [Google Scholar] [CrossRef]
- Dwivedi, B.; Xue, B.; Lundin, D.; Edwards, R.; Breitbart, M. A bioinformatic analysis of ribonucleotide reductase genes in phage genomes and metagenomes. BMC Evol. Biol. 2013, 13, 1–17. [Google Scholar] [CrossRef]
- Kelly, L.; Ding, H.; Huang, K.; Osburne, M.; Chisholm, S. Genetic diversity in cultured and wild marine cyanomyoviruses reveals phosphorus stress as a strong selective agent. ISME J. 2013, 7, 1827–1841. [Google Scholar] [CrossRef]
- Millard, A.; Clokie, M.R.J.; Shub, D.A.; Mann, N.H. Genetic organization of the psbAD region in phages infecting marine Synechococcus strains. Proc. Nat. Acad. Sci. USA 2004, 101, 11007–11012. [Google Scholar] [CrossRef]
- Bryan, M.J.; Burroughs, N.J.; Spence, E.M.; Clokie, M.R.; Mann, N.H.; Bryan, S.J. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS ONE 2008, 3, e2048. [Google Scholar] [CrossRef]
- Goldsmith, D.B.; Crosti, G.; Dwivedi, B.; McDaniel, L.D.; Varsani, A.; Suttle, C.A.; Breitbart, M. Development of phoH as a novel signature gene for assessing marine phage diversity. Appl. Environ. Microbiol. 2011, 77, 7730–7739. [Google Scholar] [CrossRef]
- Cordero, O.; Polz, M. Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 2014, 12, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lindell, D.; Jaffe, J.D.; Coleman, M.L.; Futschik, M.E.; Axmann, I.M.; Rector, T.; Kettler, G.; Sullivan, M.B.; Steen, R.; Hess, W.R.; et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature 2007, 449, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.; Benkő, M. Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Gao, C.; Jiang, Y.; Wang, M.; Zhou, X.; Shao, H.; Gong, Z.; McMinn, A. Metagenomic Characterization of the Viral Community of the South Scotia Ridge. Viruses 2019, 11, 95. [Google Scholar] [CrossRef]
- Jiang, T.; Guo, C.; Wang, M.; Wang, M.; You, S.; Liu, Y.; McMinn, A. Isolation and complete genome sequence of a novel cyanophage, S-B05, infecting an estuarine Synechococcus strain: Insights into environmental adaptation. Arch. Virol. 2020, 165, 1397–1407. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Carberry, M.J.; Eldridge, M.L.; Poorvin, L.; Saxton, M.A.; Doblin, M.A. Marine and freshwater cyanophages in a Laurentian Great Lake: Evidence from infectivity assays and molecular analyses of g20 genes. Appl. Environ. Microbiol. 2006, 72, 4957–4963. [Google Scholar] [CrossRef]
- Wilson, W.H.; Joint, I.R.; Carr, N.G.; Mann, N.H. Isolation and molecular characterization of five marine cyanophages propagated on Synechococcus sp. Strain WH7803. Appl. Environ. Microbiol. 1993, 59, 3736–3743. [Google Scholar] [CrossRef]
- Chénard, C.; Sandra, K.; Lauro, F.M. Complete genome sequence of the cyanophage S-PRM1 isolated from Singapore coastal waters. Mar. Genom. 2018, 43, 58–60. [Google Scholar] [CrossRef]
- Deveau, H.; Labrie, S.J.; Chopin, M.C.; Moineau, S. Biodiversity and classification of lactococcal phages. Appl. Environ. Microbiol. 2006, 72, 4338–4346. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Pyshkin, A.V. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Wang, M.; Jiang, Y.; Jiang, T.; Liu, Y.; Liu, X.; Shao, H. The Genome Sequence of a Novel Cyanophage S-B64 from the Yellow Sea, China. Curr. Microbiol. 2009, 76, 681–686. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Zagnitko, O. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov (accessed on 1 February 2020).
- Blast. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 1 February 2020).
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Gough, J. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef]
- CDD. Available online: https://www.ncbi.nlm.nih.gov/cdd (accessed on 1 February 2020).
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; Yandava, C. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef]
- ViPTree. Available online: https://www.genome.jp/viptree (accessed on 1 February 2020).
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Virus-Host-DB. Available online: https://www.genome.jp/virushostdb (accessed on 1 February 2020).
- Zuckerkandl, E.; Pauling, L. Evolutionary Divergence and Convergence in Proteins. In Evolving Genes and Proteins; Elsevier: Amsterdam, The Netherlands, 1965; pp. 97–166. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evolut. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evolut. Microbio. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Vidyarathna, N.K.; Palenik, B.; Lee, P.; Liu, H. Comparison of the seasonal variations of Synechococcus Assemblage structures in estuarine waters and coastal waters of Hong Kong. Appl. Environ. Microbiol. 2015, 81, 7644. [Google Scholar] [CrossRef]
- Rocha, E.P.C.; Danchin, A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002, 18, 291–294. [Google Scholar] [CrossRef]
- Almpanis, A.; Swain, M.; Gatherer, D.; McEwan, N. Correlation between bacterial G+C content, genome size and the G+C content of associated plasmids and bacteriophages. Microb. Genom. 2018, 4, e000168. [Google Scholar] [CrossRef]
- Enav, H.; Béjà, O.; Mandel-Gutfreund, Y. Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. ISME J. 2012, 6, 619–628. [Google Scholar] [CrossRef]
- Nishida, H. Evolution of genome base composition and genome size in bacteria. Front. Microbiol. 2012, 3, 420. [Google Scholar] [CrossRef]
- Mann, N.H.; Clokie, M.R.J.; Millard, A.; Cook, A.; Wilson, W.H.; Wheatley, P.J.; Letarov, A.; Krisch, H.M. The Genome of S-PM2, a “Photosynthetic” T4-Type Bacteriophage That Infects Marine Synechococcus Strains. J. Bacteriol. 2005, 187, 3188–3200. [Google Scholar] [CrossRef] [PubMed]
- Weigele, P.R.; Pope, W.H.; Pedulla, M.L.; Houtz, J.M.; Smith, A.L.; Conway, J.F.; King, J.; Hatfull, G.F.; Lawrence, J.G.; Hendrix, R.W. Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ. Microbiol. 2007, 9, 1675–1695. [Google Scholar] [CrossRef] [PubMed]
- Millard, A.D.; Zwirglmaier, K.; Downey, M.J.; Mann, N.H.; Scanlan, D.J. Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: Implications for mechanisms of cyanophage evolution. Environ. Microbiol. 2009, 11, 2370–2387. [Google Scholar] [CrossRef] [PubMed]
- Dreher, T.W.; Brown, N.; Bozarth, C.S.; Schwartz, A.D.; Riscoe, E.; Thrash, C. A freshwater cyanophage whose genome indicates close relationships to photosynthetic marine cyanomyophages. Environ. Microbiol. 2011, 13, 1858–1874. [Google Scholar] [CrossRef] [PubMed]
- Marston, M.F.; Pierciey, F.J.; Shepard, A.; Gearin, G.; Qi, J.; Yandava, J.; Schuster, S.C.; Henn, M.R.; Martiny, J.B.H. Rapid diversification of coevolving marine Synechococcus and a virus. Proc. Nat. Acad. Sci. USA 2012, 109, 4544–4549. [Google Scholar] [CrossRef] [PubMed]
- Sabehi, G.; Shaulov, L.; Silver, D.H.; Yanai, I.; Harel, A.; Lindell, D. A novel lineage of myoviruses infecting cyanobacteria is widespread in the oceans. Proc. Nat. Acad. Sci. USA 2012, 109, 2037–2042. [Google Scholar] [CrossRef]
- Deng, L.; Ignacio-Espinoza, J.C.; Gregory, A.C.; Poulos, B.T.; Weitz, J.S.; Hugenholtz, P.; Sullivan, M.B. Viral tagging reveals discrete populations in Synechococcus viral genome sequence space. Nature 2014, 513, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Solonenko, S.A.; Ignacio-Espinoza, J.C.; LaButti, K.; Copeland, A.; Sudek, S.; Maitland, A.; Chittick, L.; Santos, F.; Weitz, J.S.; et al. Genomic differentiation among wild cyanophages despite widespread horizontal gene transfer. BMC Genom. 2016, 17, 1–13. [Google Scholar] [CrossRef]
- Marston, M.F.; Martiny, J.B.H. Genomic diversification of marine cyanophages into stable ecotypes. Environ. Microbiol. 2016, 18, 4240–4253. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, R.; Wang, N.; Cai, L.; Tong, Y.; Sun, Q.; Chen, F.; Jiao, N. Novel phage–host interactions and evolution as revealed by a cyanomyovirus isolated from an estuarine environment. Environ. Microbiol. 2018, 20, 2974–2989. [Google Scholar] [CrossRef]
- Fridman, S.; Flores-Uribe, J.; Larom, S.; Alalouf, O.; Liran, O.; Yacoby, I.; Salama, F.; Bailleul, B.; Rappaport, F.; Ziv, T.; et al. A myovirus encoding both photosystem I and II proteins enhances cyclic electron flow in infected Prochlorococcus cells. Nat. Microbiol. 2017, 2, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, M.; Meng, X.; Li, Y.; Wang, D.; Jiang, Y.; Zhang, Y. Isolation and Genome Sequencing of a Novel Pseudoalteromonas Phage PH1. Curr. Microbiol. 2017, 74, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Kang, D.; Cho, J.-C. Complete genome sequence of bacteriophage P2559Y, a marine phage that infects Croceibacter atlanticus HTCC2559. Mar. Genom. 2016, 29, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Z.; Wang, M.; Liu, X.; Wang, Q.; Liu, Q.; Jiang, Y.; Li, Z.; Shao, H.; McMinn, A. Isolation and genome sequencing of the novel marine phage phs3 from the yellow sea, china. Mar. Genom. 2019, 44, 70–73. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, L.; Ma, R.; Xu, Y.; Tong, Y.; Huang, Y.; Jiao, N.; Zhang, R. A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea. Viruses 2017, 9, 109. [Google Scholar] [CrossRef]
- Stubbe, J. Ribonucleotide reductases: The link between an RNA and a DNA world? Curr. Opin. Struct. Biol. 2001, 10, 731–736. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Coleman, M.L.; Weigele, P.; Rohwer, F.; Chisholm, S.W. Three Prochlorococcus cyanophage genomes: Signature features and ecological interpretations. PLoS Biol. 2005, 3, e144. [Google Scholar] [CrossRef]
- Chen, F.; Lu, J. Genomic sequence and evolution of marine cyanophage P60: A new insight on lytic and lysogenic phages. Appl. Environ. Microbiol. 2002, 68, 2589–2594. [Google Scholar] [CrossRef]
- Mathews, C.K. An overview of the T4 Developmental Program. In Molecular Biology of Bacteriophage T4; Karam, J., Ed.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 1–8. [Google Scholar]
- Ohlsen, K.; Donat, S. The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 137–141. [Google Scholar] [CrossRef]
- Squeglia, F.; Marchetti, R.; Ruggiero, A.; Lanzetta, R.; Marasco, D.; Dworkin, J.; Petoukhov, M.; Molinaro, A.; Berisio, R.; Silipo, A. Chemical basis of peptidoglycan discrimination by PrkC, a key kinase involved in bacterial resuscitation from dormancy. J. Am. Chem. Soc. 2011, 133, 20676–20679. [Google Scholar] [CrossRef]
- Kumar, D.; Narayanan, S. pknE, a serine/threonine kinase of Mycobacterium tuberculosis modulates multiple apoptotic paradigms. Infect. Genet. Evolut. 2012, 12, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.; Campbell, K.; McAuliffe, O. Bacteriophages and Rapid Detection of Bacterial Pathogens: A Novel Approach. Ref. Modul. Life Sci. 2019, 458–466. [Google Scholar] [CrossRef]
- Krakauer, D.C.; Jansen, V.A. Red queen dynamics of protein translation. J. Theor. Biol. 2002, 218, 97–109. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef]
- Savalia, S.; Westblade, L.F.; Goel, M.; Florens, L.; Kemp, P.; Akulenko, N.; Pavlova, P.; Padovan, J.C.; Chait, B.T.; Washburn, M.P.; et al. Genomic and proteomic analysis of phiEco32, a novel Escherichia coli bacteriophage. J. Mol. Biol. 2008, 377, 774–789. [Google Scholar] [CrossRef]
- E-Bin, G.; De-Gang, N. Advances in researches on cyanophage auxiliary metabolic genes. Microbiol. China 2014, 41, 1667–1674. [Google Scholar]
- Clokie, M.R.J.; Mann, N.H. Marine cyanophages and light. Environ. Microbiol. 2006, 8, 2074–2082. [Google Scholar] [CrossRef]
- Rihtman, B.; Bowman-Grahl, S.; Millard, A.; Corrigan, R.M.; Clokie, M.R.J.; Scanlan, D.J. Cyanophage MazG is a pyrophosphohydrolase but unable to hydrolyse magic spot nucleotides. Environ. Microbiol. Rep. 2019, 11, 448–455. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Wang, X.; Liu, J.; Wang, G. Research Progress of New Biomarker Gene of phoH for Bacteriophage Genetic Diversity. Biotechnol. Bull. 2017, 33, 40–45. [Google Scholar] [CrossRef]
- Xu, J.; Glibert, P.M.; Liu, H.; Yin, K.; Yuan, X.; Chen, M.; Harrison, P.J. Nitrogen Sources and Rates of Phytoplankton Uptake in Different Regions of Hong Kong Waters in Summer. Estuar. Coasts 2012, 35, 559–571. [Google Scholar] [CrossRef]
- Hendrick, J.P.; Hartl, F.U. Molecular chaperone functions of heat-shock proteins. Annu. Rev. Biochem. 1993, 62, 349–384. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.P.; Polazzi, J.O.; Gierse, J.K.; Easton, A.M. Two novel heat shock genes encoding proteins produced in response to heterologous protein expression in Escherichia coli. J. Bacteriol. 1992, 174, 6938–6947. [Google Scholar] [CrossRef] [PubMed]
- Maaroufi, H.; Tanguay, R.M. Analysis and Phylogeny of Small Heat Shock Proteins from Marine Viruses and Their Cyanobacteria Host. PLoS ONE 2013, 8, e81207. [Google Scholar] [CrossRef]
- Bailey, S.; Melis, A.; Mackey, K.R.; Cardol, P.; Finazzi, G.; Van Dijken, G.; Berg, G.M.; Arrigo, K.; Shrager, J.; Grossman, A. Alternative photosynthetic electron flow to oxygen in marine Synechococcus. Biochim. Biophys. Acta 2008, 1777, 269–276. [Google Scholar] [CrossRef]
- McDonald, A.E.; Vanlerberghe, G.C. Alternative oxidase and plastoquinol terminal oxidase in marine prokaryotes of the Sargasso Sea. Gene 2005, 349, 15–24. [Google Scholar] [CrossRef]
- Adir, N.; Zer, H.; Shochat, S.; Ohad, I. Photoinhibition–a historical perspective. Photosynth. Res. 2003, 76, 343. [Google Scholar] [CrossRef]
- Havaux, M.; Guedeney, G.; He, Q.; Grossman, A.R. Elimination of high-light-inducible polypeptides related to eukaryotic chlorophyll a/b-binding proteins results in aberrant photoacclimation in Synechocystis PCC6803. Biochim. Biophys. Acta Bioenerg. 2003, 1557, 21–33. [Google Scholar] [CrossRef]
- Hellweger, F.L. Carrying photosynthesis genes increases ecological fitness of cyanophage in silico. Environ. Microbiol. 2009, 11, 1386–1394. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Lindell, D.; Lee, J.A.; Thompson, L.R.; Bielawski, J.P.; Chisholm, S.W. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006, 4, e234. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host | Phage | Genome Size (bp) | GC (%) | AMG | tRNA | NCBI Taxonomy ID | No. Isolates | Accession | RefSeq Accession | Ref. | Submission Date |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Synechococcus | S-PM2 | 196,280 | 37.8 | 10 | 25 | 238854 | 2 | AJ630128.1 | NC_006820.1 | [58] | 2004 |
| Syn9 | 177,300 | 40.6 | 18 | 6 | 382359 | 1 | DQ149023.2 | NC_008296.2 | [59] | 2005 | |
| S-RSM4 | 194,454 | 41.1 | 18 | 12 | 555387 | 1 | FM207411.1 | NC_013085.1 | [60] | 2008 | |
| Syn1 | 191,195 | 40.6 | 14 | 6 | 444861 | 1 | GU071105.1 | NC_015288.1 | [45] | 2009 | |
| Syn19 | 175,230 | 41.3 | 18 | 6 | 445684 | 1 | GU071106.1 | NC_015286.1 | [45] | 2009 | |
| Syn33 | 174,285 | 39.6 | 16 | 5 | 444878 | 1 | GU071108.1 | NC_015285.1 | [45] | 2009 | |
| S-SM1 | 174,079 | 41.1 | 22 | 6 | 444859 | 1 | GU071094.1 | NC_015282.1 | [45] | 2009 | |
| S-SM2 | 190,789 | 40.4 | 22 | 11 | 444860 | 1 | GU071095.1 | NC_015279.1 | [45] | 2009 | |
| S-ShM2 | 179,563 | 41.1 | 14 | 1 | 445683 | 1 | GU071096.1 | NC_015281.1 | [45] | 2009 | |
| S-SSM5 | 176,184 | 40 | 21 | 4 | 445685 | 1 | GU071097.1 | NC_015289.1 | [45] | 2009 | |
| S-SSM7 | 232,878 | 39.1 | 21 | 5 | 445686 | 1 | GU071098.1 | NC_015287.1 | [45] | 2009 | |
| Syn2 | 175,596 | 41.3 | 18 | 6 | 536473 | 1 | HQ634190.1 | - | - | 2010 | |
| Syn10 | 177,103 | 40.6 | 17 | 6 | 536472 | 1 | HQ634191.1 | - | - | 2010 | |
| Syn30 | 178,807 | 39.9 | 20 | 6 | 536474 | 1 | HQ634189.1 | NC_021072.1 | - | 2010 | |
| S-SSM2 | 179,980 | 41.1 | 14 | 1 | 536464 | 1 | JF974292.1 | - | - | 2010 | |
| S-SSM4 | 182,801 | 39.4 | 19 | 3 | 536466 | 1 | HQ316583.1 | NC_020875.1 | - | 2010 | |
| S-SSM6a | 232,883 | 39.1 | 20 | 5 | 682650 | 1 | HQ317391.1 | - | - | 2010 | |
| S-SSM6b | 182,368 | 39.4 | 19 | 3 | 682651 | 1 | HQ316603.1 | - | - | 2010 | |
| S-CAM1 | 198,013 | 43 | 17 | 8 | 754037 | 6 | HQ634177.1 | NC_020837.1 | - | 2010 | |
| S-CAM8 | 171,407 | 39.3 | 19 | 5 | 754038 | 2 | HQ634178.1 | NC_021530.1 | - | 2010 | |
| S-CRM01 | 178,563 | 39.7 | 7 | 34 | 1026955 | 1 | HQ615693.1 | NC_015569.1 | [61] | 2010 | |
| S-RIM8 | 171,211 | 40.6 | 16 | 8 | 869724 | 13 | JF974288.1 | NC_020486.1 | [62] | 2010 | |
| S-SKS1 | 208,007 | 36 | 16 | 12 | 754042 | 1 | HQ633071.1 | NC_020851.1 | - | 2010 | |
| KBS-M-1A | 171,744 | 40.6 | 16 | 8 | 889950 | 1 | JF974293.1 | - | - | 2010 | |
| S-IOM18 | 171,797 | 40.6 | 15 | 7 | 754039 | 1 | HQ317383.1 | NC_021536.1 | - | 2010 | |
| metaG-MbCM1 | 172,879 | 39.8 | 18 | 2 | 1079999 | 1 | JN371769.1 | NC_019443.1 | - | 2010 | |
| S-TIM5 | 161,440 | 40.5 | 15 | 10 | 1137745 | 1 | JQ245707.1 | NC_019516.1 | [63] | 2011 | |
| ACG-2014c | 176,043 | 39.1 | 20 | 4 | 1079998 | 5 | JN371768.1 | NC_019444.1 | [64] | 2011 | |
| ACG-2014a | 171,282 | 39.4 | 18 | 5 | 1493507 | 24 | KJ019026.1 | - | [65] | 2013 | |
| ACG-2014b | 172,688 | 39.1 | 19 | 5 | 1493508 | 18 | KJ019134.1 | NC_027130.1 | [65] | 2013 | |
| ACG-2014d | 179,110 | 40.3 | 18 | 3 | 1493509 | 45 | KJ019136.1 | NC_026923.1 | [65] | 2013 | |
| ACG-2014e | 189,418 | 38.9 | 17 | 8 | 1493510 | 3 | KJ019156.1 | NC_026928.1 | [65] | 2013 | |
| ACG-2014f | 228,143 | 41.6 | 13 | 2 | 1493511 | 41 | KJ019059.1 | NC_026927.1 | [65] | 2013 | |
| ACG-2014g | 174,885 | 39.3 | 17 | 5 | 1493512 | 1 | KJ019071.1 | NC_026924.1 | - | 2013 | |
| ACG-2014h | 189,311 | 40.5 | 16 | 7 | 1340810 | 1 | KF156338.1 | NC_023587.1 | [64] | 2013 | |
| ACG-2014i | 190,768 | 39 | 16 | 8 | 1493513 | 1 | KJ019082.1 | NC_027132.1 | [65] | 2013 | |
| ACG-2014j | 192,108 | 38.6 | 16 | 7 | 1493514 | 2 | KJ019069.1 | NC_026926.1 | [65] | 2013 | |
| S-MbCM25 | 176,044 | 39.1 | 20 | 4 | 1340811 | 1 | KF156339.1 | - | [64] | 2013 | |
| S-MbCM100 | 170,438 | 39.4 | 17 | 5 | 1340812 | 1 | KF156340.1 | NC_023584.1 | [64] | 2013 | |
| S-RIM2 | 175,430 | 42.2 | 14 | 6 | 869662 | 62 | HQ317292.1 | NC_020859.1 | [66] | 2016 | |
| S-RIM12 | 173,821 | 39.6 | 17 | 5 | 1278402 | 21 | KX349307.1 | - | [66] | 2016 | |
| S-RIM14 | 179,558 | 41.1 | 14 | 1 | 1278423 | 9 | KX349298.1 | - | [66] | 2016 | |
| S-RIM32 | 194,437 | 39.9 | 14 | 11 | 1278479 | 1 | KU594606.1 | NC_031235.1 | [11] | 2016 | |
| S-RIM44 | 197,629 | 40.3 | 19 | 5 | 1278485 | 8 | KX349291.1 | - | [66] | 2016 | |
| S-RIM50 | 174,307 | 40.3 | 16 | 8 | 687803 | 1 | KU594605.1 | NC_031242.1 | [11] | 2016 | |
| S-CAM3 | 198,190 | 41.6 | 14 | 10 | 1883366 | 3 | KU686199.1 | NC_031906.1 | [11] | 2016 | |
| S-CAM4 | 191,983 | 38.6 | 16 | 8 | 1883367 | 3 | KU686201.1 | NC_031900.1 | [11] | 2016 | |
| S-CAM7 | 216,121 | 41.2 | 5 | 4 | 1883368 | 2 | KU686212.1 | NC_031927.1 | [11] | 2016 | |
| S-CAM9 | 174,830 | 39 | 16 | 8 | 1883369 | 3 | KU686206.1 | NC_031922.1 | [11] | 2016 | |
| S-CAM22 | 172,345 | 39.9 | 20 | 5 | 1883365 | 3 | KU686209.1 | NC_031903.1 | [11] | 2016 | |
| S-WAM1 | 185,102 | 44.7 | 15 | 4 | 1815521 | 1 | KU686210.1 | NC_031944.1 | [11] | 2016 | |
| S-WAM2 | 186,386 | 41.3 | 15 | 12 | 1815522 | 1 | KU686211.1 | NC_031935.1 | [11] | 2016 | |
| S-CBWM1 | 139,069 | 51.6 | 3 | 36 | 2053653 | 1 | MG450654.1 | - | [67] | 2017 | |
| Bellamy | 204,930 | 41.1 | 20 | 10 | 2023996 | 1 | MF351863.1 | - | - | 2017 | |
| S-H35 | 174,231 | 41.2 | 15 | 8 | 1983572 | 1 | KY945241.1 | - | - | 2017 | |
| S-B68 | 163,982 | 51.7 | 4 | 4 | 2545437 | 1 | MK016664.1 | - | - | 2018 | |
| S-B64 | 151,867 | 41.3 | 15 | 8 | 2163901 | 1 | MH107246.1 | - | - | 2018 | |
| S-PRM1 | 144,311 | 40.7 | 16 | 8 | 2100130 | 1 | MH629685.1 | - | - | 2018 | |
| S-T4 | 181,082 | 38.9 | 14 | 7 | 2268578 | 1 | MH412654.1 | - | - | 2018 | |
| S-E7 | 177,622 | 39.9 | 19 | 6 | 2484639 | 1 | MH920640.1 | - | - | 2018 | |
| B3 | 244,930 | 35.4 | 4 | 20 | 2674978 | 1 | MN695334.1 | - | - | 2019 | |
| B23 | 243,633 | 35.4 | 4 | 20 | 2674977 | 1 | MN695335.1 | - | - | 2019 | |
| S-RIM4 | 175,462 | 41.2 | 15 | 9 | 2530169 | 1 | MK493321.1 | - | - | 2019 | |
| S-B43 | 213,993 | 39.4 | 14 | 11 | 1340812 | 1 | MN018232.1 | - | - | 2019 | |
| S-B05 | 208,857 | 39.9 | 14 | 11 | 2484637 | 1 | MK799832.1 | - | [27] | 2019 | |
| S-H34 | 167,040 | 50.1 | 3 | 5 | 2718942 | 1 | MT162467.2 | - | This study | 2020 | |
| S-N03 | 167,069 | 50.1 | 4 | 1 | 2718943 | 1 | MT162466.1 | - | This study | 2020 | |
| Prochlorococcus | P-SSM2 | 252,401 | 35.5 | 22 | 1 | 268746 | 2 | AY939844.2 | NC_006883.2 | [45] | 2005 |
| P-SSM4 | 178,249 | 36.7 | 19 | 0 | 268747 | 1 | AY940168.2 | NC_006884.2 | [45] | 2005 | |
| P-SSM7 | 182,180 | 37.1 | 19 | 4 | 445688 | 1 | GU071103.1 | NC_015290.1 | [45] | 2005 | |
| P-RSM4 | 176,428 | 37.6 | 19 | 3 | 444862 | 1 | GU071099.1 | NC_015283.1 | [45] | 2009 | |
| P-HM1 | 181,044 | 37.8 | 17 | 0 | 445700 | 1 | GU071101.1 | NC_015280.1 | [45] | 2009 | |
| P-HM2 | 183,806 | 38.1 | 15 | 0 | 445696 | 1 | GU075905.1 | NC_015284.1 | [45] | 2009 | |
| P-RSM1 | 177,211 | 40.2 | 18 | 2 | 536444 | 1 | HQ634175.1 | NC_021071.1 | - | 2010 | |
| P-RSM3 | 178,750 | 36.7 | 17 | 0 | 536446 | 1 | HQ634176.1 | - | - | 2010 | |
| P-RSM6 | 192,497 | 39.3 | 18 | 3 | 929832 | 1 | HQ634193.1 | NC_020855.1 | - | 2010 | |
| P-SSM3 | 179,063 | 36.7 | 16 | 0 | 536453 | 1 | HQ337021.1 | NC_021559.1 | - | 2010 | |
| P-SSM5 | 252,013 | 35.5 | 19 | 1 | 536454 | 1 | HQ632825.1 | - | - | 2010 | |
| MED4–213 | 180,977 | 37.8 | 15 | 0 | 889956 | 1 | HQ634174.1 | NC_020845.1 | - | 2010 | |
| P-TIM40 | 188,632 | 40.7 | 17 | 1 | 1589733 | 1 | KP211958.1 | NC_028663.1 | - | 2014 | |
| P-TIM68 | 197,361 | 34.3 | 14 | 0 | 1542477 | 1 | KM359505.1 | NC_028955.1 | [68] | 2014 |
| Genome Size | GC% | AMGs | ||
|---|---|---|---|---|
| Genome size | Correlation coefficient | 1 | −0.340 ** | 0.001 |
| Sig.(p-value) | - | 0.002 | 0.992 | |
| GC% | Correlation coefficient | −0.340 ** | 1 | −0.272 * |
| Sig.(p-value) | 0.002 | - | 0.014 | |
| AMGs | Correlation coefficient | 0.001 | −0.272 * | 1 |
| Sig.(p-value) | 0.992 | 0.014 | - |
| Sequence Name | Position | Length (nt) | tRNA Type | Anticodon | Isotype Model | Isotype Score |
|---|---|---|---|---|---|---|
| Synechococcus phage S-N03 | ||||||
| tRNA1 | 101128–101057 | 72 | Asn | GTT | Ans | 98.7 |
| Synechococcus phage S-H34 | ||||||
| tRNA1 | 157447–157366 | 82 | Tyr | GTA | Tyr | 70.1 |
| tRNA2 | 155156–155085 | 72 | Asn | GTT | Ans | 98.7 |
| tRNA3 | 155081–155009 | 73 | Asp | GTC | Asp | 66.5 |
| tRNA4 | 154982–154911 | 72 | Asn | GTT | Ans | 98.7 |
| tRNA5 | 154812–154741 | 72 | Val | TAC | Phe | 77.6 |
| Phage | AMGs | Genome Size (kb) | GC (%) | Isolation Location | Host Name (Syn) | Host Isolation | Accession |
|---|---|---|---|---|---|---|---|
| S-N03 | 3 | 167,069 | 50.1 | Yellow Sea, China | MW02 | estuary | MT162466 |
| S-H34 | 3 | 167,040 | 50.1 | Yellow Sea, China | MW02 | estuary | MT162467.2 |
| S-B68 | 4 | 163,982 | 51.7 | Bohai Sea, China | WH7803 | marine | MK016664.1 |
| S-CBWM1 | 4 | 139,069 | 51.6 | Chesapeake Bay, USA | CBW1002 | estuary | MG450654.1 |
| S-CAM7 | 5 | 216,121 | 41.2 | Crystal Cove, CA | WH7803 | marine | NC_031927.1 |
| S-CRM01 | 7 | 178,563 | 39.7 | Copco Reservoir, Klamath River, CA | LC16 | freshwater | NC_015569.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, T.; Guo, C.; Wang, M.; Wang, M.; Zhang, X.; Liu, Y.; Liang, Y.; Jiang, Y.; He, H.; Shao, H.; et al. Genome Analysis of Two Novel Synechococcus Phages That Lack Common Auxiliary Metabolic Genes: Possible Reasons and Ecological Insights by Comparative Analysis of Cyanomyoviruses. Viruses 2020, 12, 800. https://doi.org/10.3390/v12080800
Jiang T, Guo C, Wang M, Wang M, Zhang X, Liu Y, Liang Y, Jiang Y, He H, Shao H, et al. Genome Analysis of Two Novel Synechococcus Phages That Lack Common Auxiliary Metabolic Genes: Possible Reasons and Ecological Insights by Comparative Analysis of Cyanomyoviruses. Viruses. 2020; 12(8):800. https://doi.org/10.3390/v12080800
Chicago/Turabian StyleJiang, Tong, Cui Guo, Min Wang, Meiwen Wang, Xinran Zhang, Yundan Liu, Yantao Liang, Yong Jiang, Hui He, Hongbing Shao, and et al. 2020. "Genome Analysis of Two Novel Synechococcus Phages That Lack Common Auxiliary Metabolic Genes: Possible Reasons and Ecological Insights by Comparative Analysis of Cyanomyoviruses" Viruses 12, no. 8: 800. https://doi.org/10.3390/v12080800
APA StyleJiang, T., Guo, C., Wang, M., Wang, M., Zhang, X., Liu, Y., Liang, Y., Jiang, Y., He, H., Shao, H., & McMinn, A. (2020). Genome Analysis of Two Novel Synechococcus Phages That Lack Common Auxiliary Metabolic Genes: Possible Reasons and Ecological Insights by Comparative Analysis of Cyanomyoviruses. Viruses, 12(8), 800. https://doi.org/10.3390/v12080800