High-Quality Resolution of the Outbreak-Related Zika Virus Genome and Discovery of New Viruses Using Ion Torrent-Based Metatranscriptomics

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture, Infection, Library Preparation and Sequencing
2.2. Reference Genomes.
2.3. Bioinformatics Analyses.
2.4. Validation of Totivirus presence by RT-PCR and Sanger Sequencing
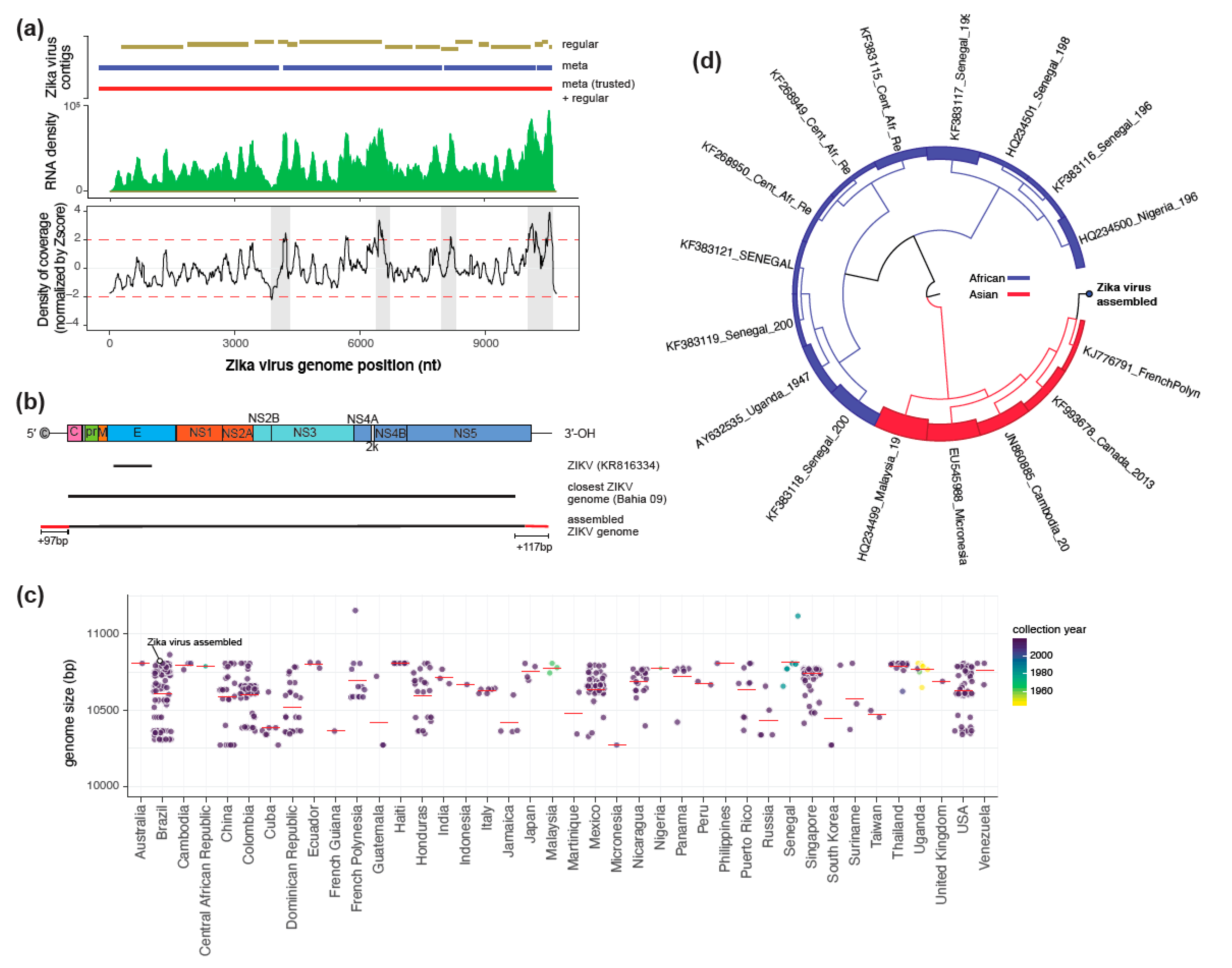
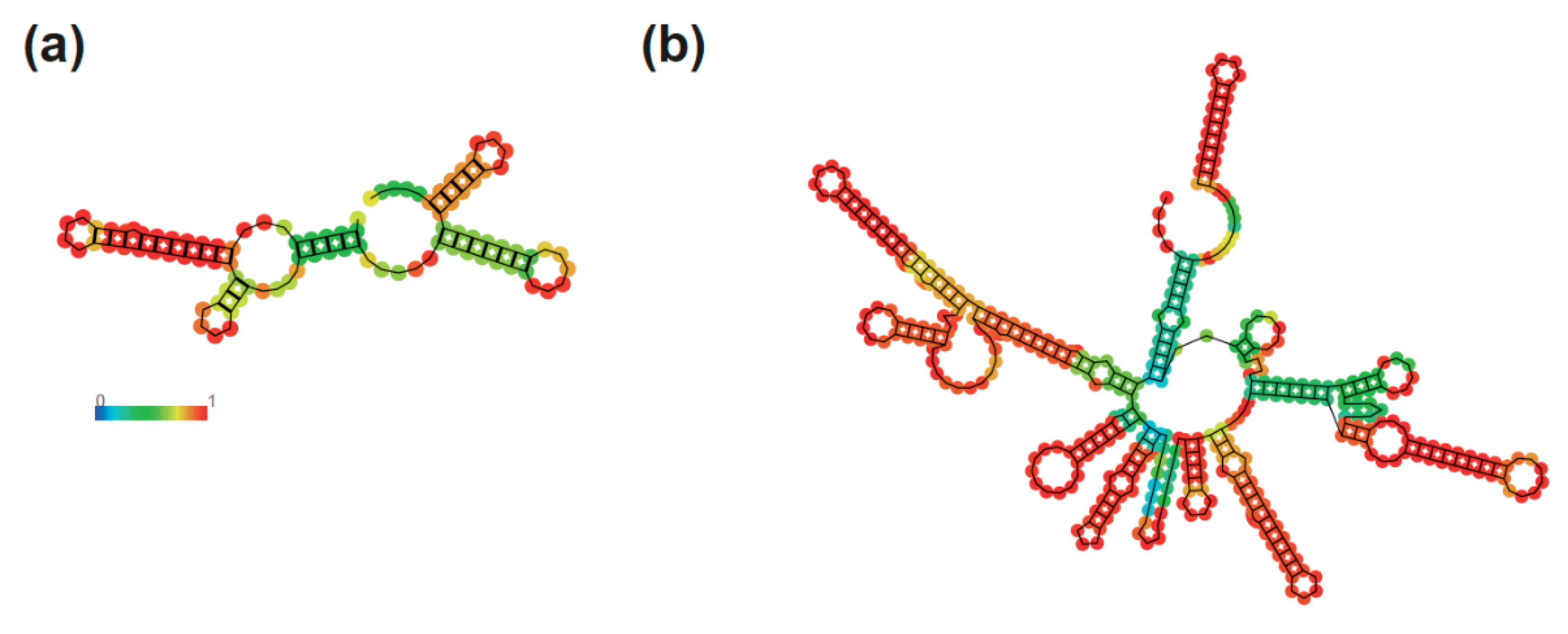
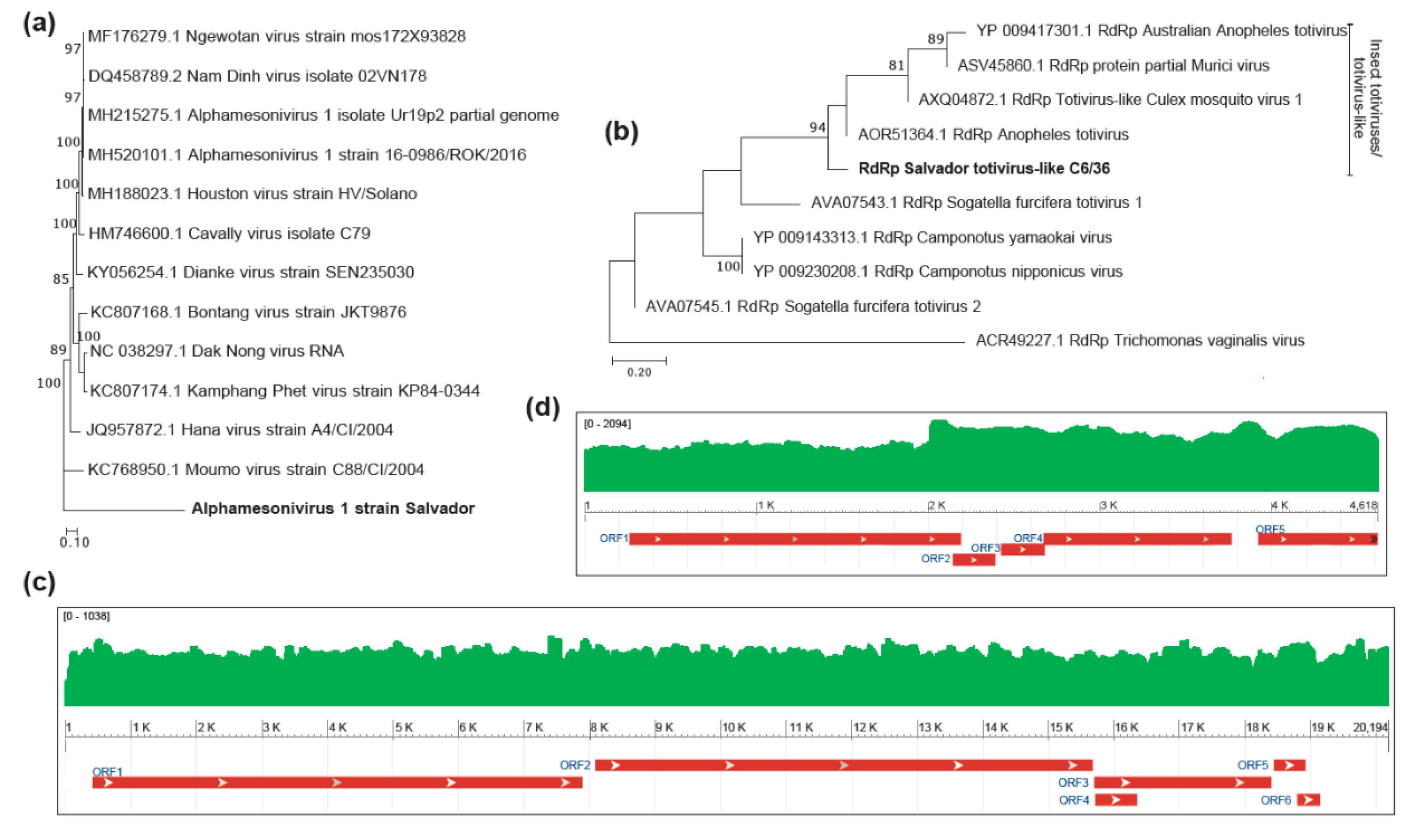
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kraemer, M.U.G.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.N.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife 2015, 4, e08347. [Google Scholar] [CrossRef] [PubMed]
- Leta, S.; Beyene, T.J.; De Clercq, E.M.; Amenu, K.; Kraemer, M.U.G.; Revie, C.W. Global risk mapping for major diseases transmitted by Aedes aegypti and Aedes albopictus. Int. J. Infect. Dis. 2018, 67, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika Virus Outbreak, Bahia, Brazil. Emerg. Infect. Dis. 2015, 21, 1885–1886. [Google Scholar] [CrossRef] [PubMed]
- Simonin, Y.; van Riel, D.; de Perre, P.V.; Rockx, B.; Salinas, S. Differential virulence between Asian and African lineages of Zika virus. PLoS Negl. Trop. Dis. 2017, 11, e0005821. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Thurmond, S.; Islas, L.; Hui, K.; Hai, R. Zika virus genome biology and molecular pathogenesis. Emerg. Microbes Infect. 2017, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chan, J.F.-W.; Tee, K.-M.; Choi, G.K.-Y.; Lau, S.K.-P.; Woo, P.C.-Y.; Tse, H.; Yuen, K.-Y. Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg. Microbes Infect. 2016, 5, e22. [Google Scholar] [CrossRef]
- Lowe, R.; Barcellos, C.; Brasil, P.; Cruz, O.G.; Honório, N.A.; Kuper, H.; Carvalho, M.S. The Zika Virus Epidemic in Brazil: From Discovery to Future Implications. Int. J. Environ. Res. Public Health 2018, 15, 96. [Google Scholar] [CrossRef]
- Aguiar, E.R.; Olmo, R.P.; Paro, S.; Ferreira, F.V.; de Faria, I.J.; Todjro, Y.M.; Lobo, F.P.; Kroon, E.G.; Meignin, C.; Gatherer, D.; et al. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res. 2015, 43, 6191–6206. [Google Scholar] [CrossRef]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rúa, A.; Failloux, A.-B.; Matthijnssens, J. Stable distinct core eukaryotic viromes in different mosquito species from Guadeloupe, using single mosquito viral metagenomics. Microbiome 2019, 7, 121. [Google Scholar] [CrossRef]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef]
- Bolling, B.G.; Olea-Popelka, F.J.; Eisen, L.; Moore, C.G.; Blair, C.D. Transmission dynamics of an insect-specific flavivirus in a naturally infected Culex pipiens laboratory colony and effects of co-infection on vector competence for West Nile virus. Virology 2012, 427, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Hall-Mendelin, S.; McLean, B.J.; Bielefeldt-Ohmann, H.; Hobson-Peters, J.; Hall, R.A.; van den Hurk, A.F. The insect-specific Palm Creek virus modulates West Nile virus infection in and transmission by Australian mosquitoes. Parasites Vectors 2016, 9, 414. [Google Scholar] [CrossRef]
- Öhlund, P.; Lundén, H.; Blomström, A.-L. Insect-specific virus evolution and potential effects on vector competence. Virus Genes 2019, 55, 127–137. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, P.; Sklan, E.; Wilkins, C.; Burgon, T.; Samuel, M.A.; Lu, R.; Ansel, K.M.; Heissmeyer, V.; Einav, S.; Jackson, W.; et al. Six RNA viruses and forty-one hosts: Viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog. 2010, 6, e1000764. [Google Scholar] [CrossRef] [PubMed]
- Vainio, E.J.; Jurvansuu, J.; Streng, J.; Rajamaki, M.-L.; Hantula, J.; Valkonen, J.P.T. Diagnosis and discovery of fungal viruses using deep sequencing of small RNAs. J. Gen. Virol. 2015, 96, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.L.C.; Badotti, F.; de Oliveira, T.F.P.; Fonseca, A.; Vaz, A.B.M.; Tomé, L.M.R.; Abrahão, J.S.; Marques, J.T.; Trindade, G.S.; Chaverri, P.; et al. Virome analyses of Hevea brasiliensis using small RNA deep sequencing and PCR techniques reveal the presence of a potential new virus. Virol. J. 2018, 15, 184. [Google Scholar] [CrossRef]
- Pop, M. Genome assembly reborn: Recent computational challenges. Brief. Bioinform. 2009, 10, 354–366. [Google Scholar] [CrossRef]
- Mangul, S.; Wu, N.C.; Mancuso, N.; Zelikovsky, A.; Sun, R.; Eskin, E. Accurate viral population assembly from ultra-deep sequencing data. Bioinformatics 2014, 30, i329–i337. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 September 2017).
- Gordon, A.; Hannon, G. Fastx-Toolkit. FASTQ/A Short-Reads Pre-Processing Tools. 2010. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 20 November 2017).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012; ISBN 3-900051-07-0. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 3-319-24277-6. [Google Scholar]
- Biology of Negative Strand RNA Viruses: The Power of Reverse Genetics; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2004; ISBN 978-3-540-40661-7.
- White, K.A.; Enjuanes, L.; Berkhout, B. RNA virus replication, transcription and recombination. RNA Biol. 2011, 8, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Brinton, M.A.; Basu, M. Functions of the 3′ and 5′ genome RNA regions of members of the genus Flavivirus. Virus Res. 2015, 206, 108–119. [Google Scholar] [CrossRef]
- Proutski, V.; Gaunt, M.W.; Gould, E.A.; Holmes, E.C. Secondary structure of the 3′-untranslated region of yellow fever virus: Implications for virulence, attenuation and vaccine development. J. Gen. Virol. 1997, 78, 1543–1549. [Google Scholar] [CrossRef]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef]
- Vasilakis, N.; Guzman, H.; Firth, C.; Forrester, N.L.; Widen, S.G.; Wood, T.G.; Rossi, S.L.; Ghedin, E.; Popov, V.; Blasdell, K.R.; et al. Mesoniviruses are mosquito-specific viruses with extensive geographic distribution and host range. Virol. J. 2014, 11, 97. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Ge, X.; Yuan, J.; Shi, Z. A novel totivirus-like virus isolated from bat guano. Arch. Virol 2012, 157, 1093–1099. [Google Scholar] [CrossRef]
- Park, Y.; James, D.; Punja, Z.K. Co-infection by two distinct totivirus-like double-stranded RNA elements in Chalara elegans (Thielaviopsis basicola). Virus Res. 2005, 109, 71–85. [Google Scholar] [CrossRef]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of > 12 thousand Culex mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Sabanadzovic, S.; Valverde, R.A.; Brown, J.K.; Martin, R.R.; Tzanetakis, I.E. Southern tomato virus: The link between the families Totiviridae and Partitiviridae. Virus Res. 2009, 140, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Lachnit, T.; Thomas, T.; Steinberg, P. Expanding our Understanding of the Seaweed Holobiont: RNA Viruses of the Red Alga Delisea pulchra. Front. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
I. Sardi, S.; H. Carvalho, R.; C. Pacheco, L.G.; P. d. Almeida, J.P.; M. d. A. Belitardo, E.M.; S. Pinheiro, C.; S. Campos, G.; R. G. R. Aguiar, E. High-Quality Resolution of the Outbreak-Related Zika Virus Genome and Discovery of New Viruses Using Ion Torrent-Based Metatranscriptomics. Viruses 2020, 12, 782. https://doi.org/10.3390/v12070782
I. Sardi S, H. Carvalho R, C. Pacheco LG, P. d. Almeida JP, M. d. A. Belitardo EM, S. Pinheiro C, S. Campos G, R. G. R. Aguiar E. High-Quality Resolution of the Outbreak-Related Zika Virus Genome and Discovery of New Viruses Using Ion Torrent-Based Metatranscriptomics. Viruses. 2020; 12(7):782. https://doi.org/10.3390/v12070782
Chicago/Turabian StyleI. Sardi, Silvia, Rejane H. Carvalho, Luis G. C. Pacheco, João P. P. d. Almeida, Emilia M. M. d. A. Belitardo, Carina S. Pinheiro, Gúbio S. Campos, and Eric R. G. R. Aguiar. 2020. "High-Quality Resolution of the Outbreak-Related Zika Virus Genome and Discovery of New Viruses Using Ion Torrent-Based Metatranscriptomics" Viruses 12, no. 7: 782. https://doi.org/10.3390/v12070782
APA StyleI. Sardi, S., H. Carvalho, R., C. Pacheco, L. G., P. d. Almeida, J. P., M. d. A. Belitardo, E. M., S. Pinheiro, C., S. Campos, G., & R. G. R. Aguiar, E. (2020). High-Quality Resolution of the Outbreak-Related Zika Virus Genome and Discovery of New Viruses Using Ion Torrent-Based Metatranscriptomics. Viruses, 12(7), 782. https://doi.org/10.3390/v12070782