Assessing Genome-Wide Diversity in European Hantaviruses through Sequence Capture from Natural Host Samples


Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Samples, RNA Extraction and Sanger Sequencing
2.2. Library Preparation
2.3. Hybrid Sequence Capture
2.4. Sequencing and Hantavirus Genome Assembly
2.5. Phylogenetic Analyses
2.6. Multidimensional Scaling, Recombination and Isolation-By-Distance Analyses
2.7. Nucleotide Diversity and Divergence between Phylogenetic Clades
3. Results
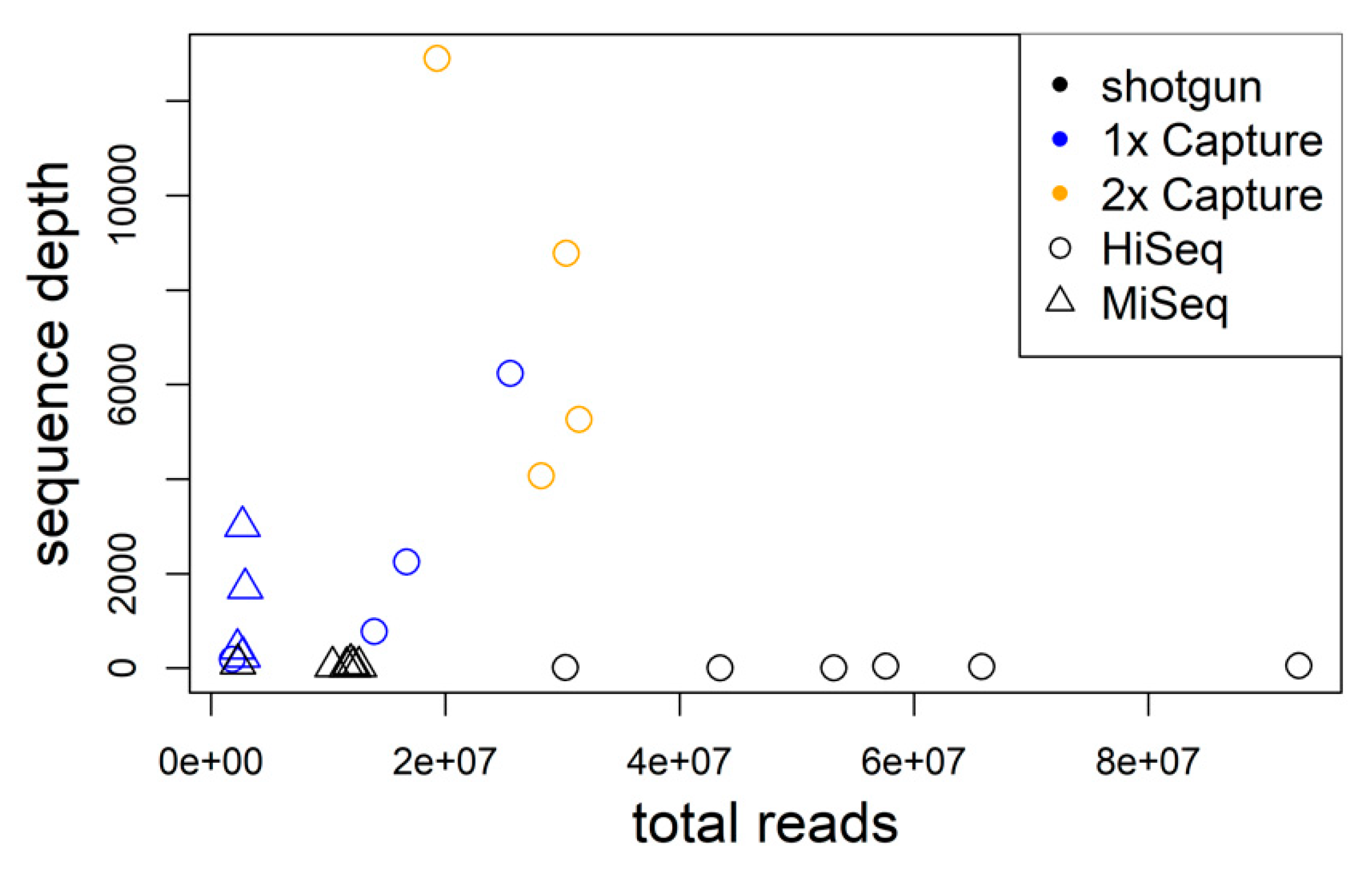
3.1. Hybrid Sequence Capture and Sequencing of TULV and PUUV Genomes
3.2. Novel Hantavirus Genome Assemblies
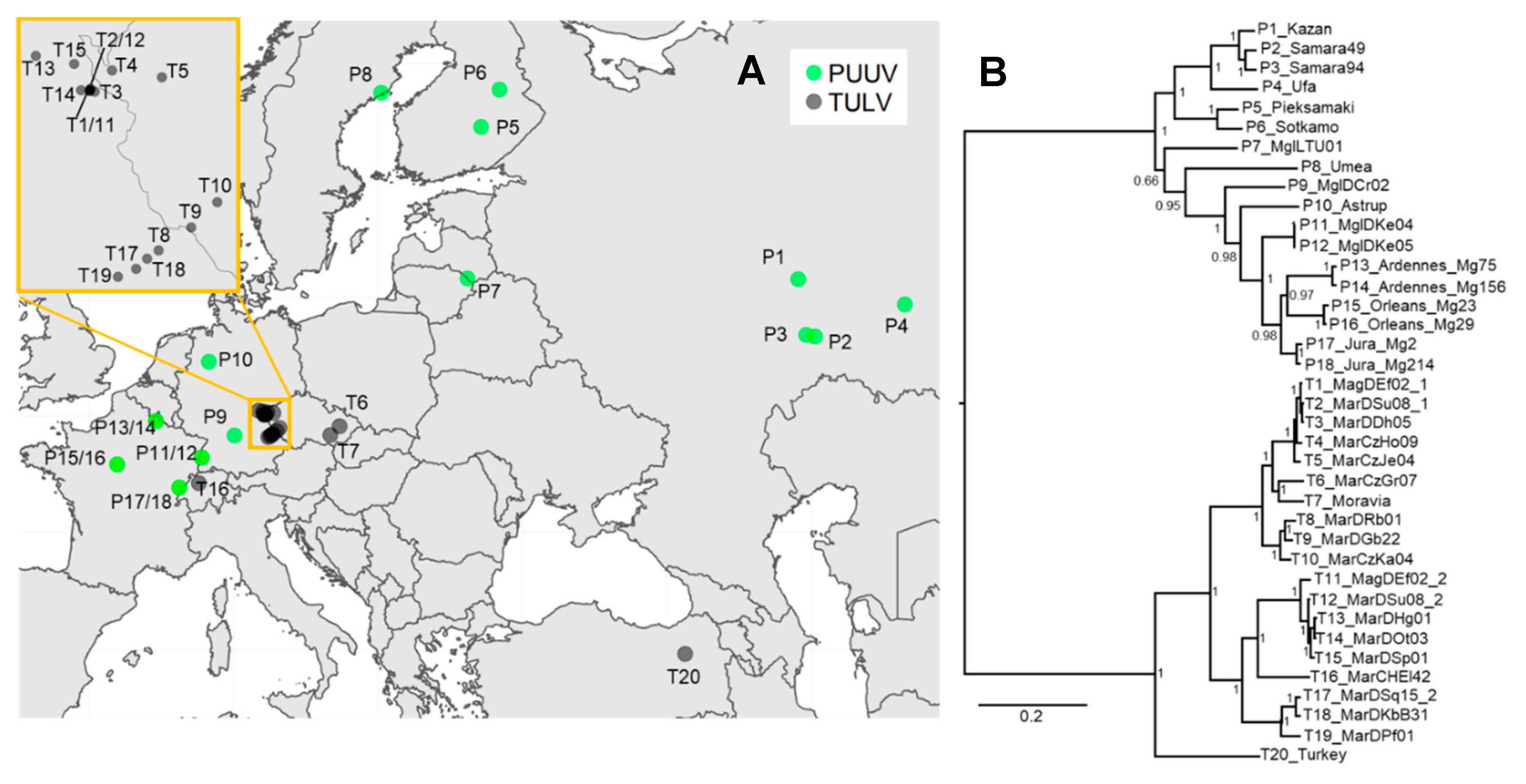
3.3. Phylogenetic, Recombination and Isolation-By-Distance Analyses
3.4. Nucleotide Diversity and Pattern along the Genome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Nemirov, K.; Vapalahti, O.; Papa, A.; Plyusnina, A.; Lundkvist, Å.; Antoniadis, A.; Vaheri, A.; Plyusnin, A. Genetic characterization of new Dobrava hantavirus isolate from Greece. J. Med. Virol. 2003, 69, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.D.; Rowe, J.E.; Borucki, M.K.; Jeor, S.C.S. Complete nucleotide sequence of a Chilean hantavirus. Virus Res. 2002, 89, 131–143. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Vanmechelen, B.; Tersago, K.; Baele, G.; Lemey, P.; Leirs, H.; Dellicour, S.; Vrancken, B.; Maes, P. Identifying the patterns and drivers of Puumala hantavirus enzootic dynamics using reservoir sampling. Virus Evol. 2019, 5, vez009. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; Drewes, S.; Weber de Melo, V.; Schlegel, M.; Freise, J.; Groschup, M.H.; Heckel, G.; Ulrich, R.G. Complete genome of a Puumala virus strain from Central Europe. Virus Genes 2015, 50, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Jeske, K.; Hiltbrunner, M.; Drewes, S.; Ryll, R.; Wenk, M.; Špakova, A.; Petraitytė-Burneikienė, R.; Heckel, G.; Ulrich, R.G. Field vole-associated Traemmersee hantavirus from Germany represents a novel hantavirus species. Virus Genes 2019, 55, 848–853. [Google Scholar] [CrossRef]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef]
- Filippone, C.; Castel, G.; Murri, S.; Ermonval, M.; Korva, M.; Avšič-Županc, T.; Sironen, T.; Vapalahati, O.; McElhinney, L.M.; Ulrich, R.G.; et al. Revisiting the genetic diversity of emerging hantaviruses circulating in Europe using a pan-viral resequencing microarray. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Polat, C.; Ergünay, K.; Irmak, S.; Erdin, M.; Brinkmann, A.; Çetintaş, O.; Çoğal, M.; Sözen, M.; Matur, F.; Nitsche, A.; et al. A novel genetic lineage of Tula orthohantavirus in Altai voles (Microtus obscurus) from Turkey. Infect. Genet. Evol. 2019, 67, 150–158. [Google Scholar] [CrossRef]
- No, J.S.; Kim, W.K.; Cho, S.; Lee, S.H.; Kim, J.A.; Lee, D.; Song, D.H.; Gu, S.H.; Jeong, S.T.; Wiley, M.R.; et al. Comparison of targeted next-generation sequencing for whole-genome sequencing of Hantaan orthohantavirus in Apodemus agrarius lung tissues. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Manso, C.F.; Bibby, D.F.; Mbisa, J.L. Efficient and unbiased metagenomic recovery of RNA virus genomes from human plasma samples. Sci. Rep. 2017, 7, 4173. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.; Rouillard, J.M.; Poinar, H. Quantitative PCR as a predictor of aligned ancient DNA read counts following targeted enrichment. BioTechniques 2013, 55, e309. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Arcos, M.C.; Sandoval-Velasco, M.; Schroeder, H.; Carpenter, M.L.; Malaspinas, A.S.; Wales, N.; Peñaloza, F.; Bustamante, C.D.; Gilbert, M.T.P. Comparative performance of two whole-genome capture methodologies on ancient DNA Illumina libraries. Methods Ecol. Evol. 2015, 6, 725–734. [Google Scholar] [CrossRef]
- Carpi, G.; Walter, K.S.; Bent, S.J.; Hoen, A.G.; Diuk-Wasser, M.; Caccone, A. Whole genome capture of vector-borne pathogens from mixed DNA samples: A case study of Borrelia burgdorferi. BMC Genom. 2015, 16, 434. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.C.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef] [PubMed]
- Burrel, S.; Boutolleau, D.; Ryu, D.; Agut, H.; Merkel, K.; Leendertz, F.H.; Calvignac-Spencer, S. Ancient recombination events between human herpes simplex viruses. Mol. Biol. Evol. 2017, 34, 1713–1721. [Google Scholar] [CrossRef]
- Gasc, C.; Peyretaillade, E.; Peyret, P. Sequence capture by hybridization to explore modern and ancient genomic diversity in model and nonmodel organisms. Nucleic Acids Res. 2016, 44, 4504–4518. [Google Scholar] [CrossRef]
- Metsky, H.C.; Siddle, K.J.; Gladden-Young, A.; Qu, J.; Yang, D.K.; Brehio, P.; Goldfarb, A.; Piantadosi, A.; Wohl, S.; Carter, A. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat. Biotechnol. 2019, 37, 160. [Google Scholar] [CrossRef]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. MBio 2015, 6. [Google Scholar] [CrossRef]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.; Melo, F.L.; Figueiredo, L.M.; Holmes, E.C.; Zanotto, P.M.; VGDN Consortium. High rates of molecular evolution in hantaviruses. Mol. Biol. Evol. 2008, 25, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Weber de Melo, V.; Sheikh Ali, H.; Freise, J.; Kuhnert, D.; Essbauer, S.S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal dynamics of Puumala hantavirus associated with its rodent host, Myodes glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Saxenhofer, M.; Weber de Melo, V.; Ulrich, R.G.; Heckel, G. Revised time scales of RNA virus evolution based on spatial information. Proc. R. Soc. B 2017, 284, 20170857. [Google Scholar] [CrossRef] [PubMed]
- Szabó, R.; Radosa, L.; Ličková, M.; Sláviková, M.; Heroldová, M.; Stanko, M.; Pejčoch, M.; Osterberg, A.; Laenen, L.; Schex, S.; et al. Phylogenetic analysis of Puumala virus strains from Central Europe highlights the need for a full-genome perspective on hantavirus evolution. Virus Genes 2017, 53, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Saxenhofer, M.; Schmidt, S.; Ulrich, R.G.; Heckel, G. Secondary contact between diverged host lineages entails ecological speciation in a European hantavirus. PLoS Biol. 2019, 17, e3000142. [Google Scholar] [CrossRef]
- Schmidt, S.; Saxenhofer, M.; Drewes, S.; Schlegel, M.; Wanka, K.M.; Frank, R.; Klimpel, S.; von Blanckenhagen, F.; Maaz, D.; Herden, C.; et al. High genetic structuring of Tula Hantavirus. Arch. Virol. 2016, 161, 1135–1149. [Google Scholar] [CrossRef]
- Schmidt-Chanasit, J.; Essbauer, S.; Petraityte, R.; Yoshimatsu, K.; Tackmann, K.; Conraths, F.J.; Sasnauskas, K.; Arikawa, J.; Thomas, A.; Pfeffer, M.; et al. Extensive host sharing of Central European Tula virus. J. Virol. 2010, 84, 459–474. [Google Scholar] [CrossRef]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2011, 40, e3. [Google Scholar] [CrossRef]
- Rohland, N.; Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012, 22, 939–946. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomics datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Gall, A.; Ong, S.H.; Brener, J.; Ferns, B.; Goulder, P.; Nastouli, E.; Keane, J.A.; Kellam, P.; Otto, T.D. IVA: Accurate de novo assembly of RNA virus genomes. Bioinformatics 2015, 31, 2374–2376. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Martin, J.; Bruno, V.M.; Fang, Z.; Meng, X.; Blow, M.; Zhang, T.; Sherlock, G.; Snyder, M.; Wang, Z. Rnnotator: An automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genom. 2010, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabrial, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; Available online: https://www.r-project.org (accessed on 3 November 2019).
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Gateway Computing Environments Workshop (GCE); IEEE: Piscataway, NJ, USA, 2010; pp. 1–8. Available online: http://www.phylo.org (accessed on 22 September 2019).
- Rambaut, A. FigTree v1.4.2. Molecular Evolution, Phylogenetics and Epidemiology; University of Edinburgh, Institute of evolutionary Biology: Edinburgh, UK, 2012. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Degner, J.F.; Marioni, J.C.; Pai, A.A.; Pickrell, J.K.; Nkadori, E.; Gilad, Y.; Pritchard, J.K. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 2009, 25, 3207–3212. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef]
- Chaisson, M.J.; Wilson, R.K.; Eichler, E.E. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015, 16, 627–640. [Google Scholar] [CrossRef]
- Burri, R.; Nater, A.; Kawakami, T.; Mugal, C.F.; Olason, P.I.; Smeds, L.; Suh, A.; Dutoit, L.; Bureš, S.; Garamszegi, L.Z.; et al. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 2015, 25, 1656–1665. [Google Scholar] [CrossRef]
- Vijay, N.; Weissensteiner, M.; Burri, R.; Kawakami, T.; Ellegren, H.; Wolf, J.B. Genome-wide patterns of variation in genetic diversity are shared among populations, species and higher order taxa. Mol. Ecol. 2017, 25, 1656–1665. [Google Scholar]
- Wolf, J.B.; Ellegren, H. Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet. 2017, 18, 87. [Google Scholar] [CrossRef]
- Han, G.Z.; Worobey, M. Homologous recombination in negative sense RNA viruses. Viruses 2011, 3, 1358–1373. [Google Scholar] [CrossRef]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Hillung, J.; Cuevas, J.M.; Elena, S.F. Evaluating the within-host fitness effects of mutations fixed during virus adaptation to different ecotypes of a new host. Philos. Trans. R. Soc. B 2015, 370, 20140292. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, S.S.; Mir, M.A. The role of viral genomic RNA and nucleocapsid protein in the autophagic clearance of hantavirus glycoprotein Gn. Virus Res. 2014, 187, 72–76. [Google Scholar] [CrossRef]
- Cheng, E.; Wang, Z.; Mir, M.A. Interaction between hantavirus nucleocapsid protein (N) and RNA-dependent RNA polymerase (RdRp) mutants reveals the requirement of an N-RdRp interaction for viral RNA synthesis. J. Virol. 2014, 88, 8706–8712. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Mir, M.A. Interaction of hantavirus nucleocapsid protein with ribosomal protein S19. J. Virol. 2010, 84, 12450–12453. [Google Scholar] [CrossRef]
- Cheng, E.; Haque, A.; Rimmer, M.A.; Hussein, I.T.; Sheema, S.; Little, A.; Mir, M.A. Characterization of the interaction between hantavirus nucleocapsid protein (N) and ribosomal protein S19 (RPS19). J. Biol. Chem. 2011, 286, 11814–11824. [Google Scholar] [CrossRef]
- Park, S.W.; Han, M.G.; Park, C.; Ju, Y.R.; Ahn, B.Y.; Ryou, J. Hantaan virus nucleocapsid protein stimulates MDM2-dependent p53 degradation. J. Gen. Virol. 2013, 94, 2424–2428. [Google Scholar] [CrossRef]
- Cimica, V.; Dalrymple, N.A.; Roth, E.; Nasonov, A.; Mackow, E.R. An innate immunity-regulating virulence determinant is uniquely encoded within the Andes virus nucleocapsid protein. MBio 2014, 5. [Google Scholar] [CrossRef]
- Jääskeläinen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef]
- Krakauer, D.C. Stability and evolution of overlapping genes. Evolution 2000, 54, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Neher, R.A. Quantifying selection against synonymous mutations in HIV-1 env evolution. J. Virol. 2013, 87, 11843–11850. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Capture | Sequencer | Total Reads | Unique Reads | Virus Reads | Mean Sequence Depth | |
|---|---|---|---|---|---|---|
| Tula virus | ||||||
| T10 MarCzKa04 | 0 | HiSeq | 30,240,364 | 30,239,770 | 1737 | 9 |
| T9 MarDGb22 | 0 | HiSeq | 43,470,283 | 43,469,619 | 1781 | 8 |
| T18 MarDKbB31 | 0 | HiSeq | 92,841,279 | 92,824,579 | 26,721 | 48 |
| T19 MarDPf01 | 0 | HiSeq | 65,793,875 | 65,787,860 | 12,006 | 34 |
| T8 MarDRb01 | 0 | HiSeq | 57,580,789 | 57,571,558 | 16,077 | 38 |
| T13 MarDHg01 | 0 | MiSeq | 11,563,256 | 11,563,254 | 1539 | 30 |
| T3 MarDDh05 | 0 | MiSeq | 12,157,472 | 12,157,461 | 1712 | 34 |
| T4 MarCzHo09 | 0 | MiSeq | 10,363,464 | 10,363,453 | 1850 | 38 |
| T5 MarCzJe04 | 0 | MiSeq | 12,634,308 | 12,634,294 | 1974 | 38 |
| T15 MarDSp01 | 0 | MiSeq | 2,286,266 | 2,286,252 | 4766 | 97 |
| T14 MarDOt03 | 0 | MiSeq | 11,907,702 | 2,286,176 | 4682 | 91 |
| T17 MarDSq15_1 1 | 0 | HiSeq | 53,177,359 | 53,177,238 | 583 | 4 |
| T17 MarDSq15_2 1 | 1 | HiSeq | 25,564,658 | 22,901,877 | 515,194 | 6238 |
| T6 MarCzGr07 | 1 | HiSeq | 13,928,878 | 10,770,028 | 62,939 | 771 |
| T2/T12 MarDSu08_1 1 | 1 | HiSeq | 16,711,898 | 12,660,735 | 184,258 | 2249 |
| T2/T12 MarDSu08_2 1 | 2 | HiSeq | 28,180,378 | 1,069,556 | 334,893 | 4068 |
| T2/T12 MarDSu08_3 1 | 1 | MiSeq | 1,812,673 | 1,812,673 | 19,035 | 378 |
| T1/T11 MagDEf02_1 1 | 2 | HiSeq | 30,333,274 | 1,168,089 | 734,578 | 8780 |
| T1/T11 MagDEf02_2 1 | 2 | HiSeq | 31,420,140 | 1,099,805 | 436,943 | 5267 |
| T16 MarCHEl42 | 2 | HiSeq | 19,306,058 | 1,666,013 | 1,066,770 | 12,896 |
| Puumala virus | ||||||
| P9 MglDCr02 | 1 | MiSeq | 2,914,826 | 1,779,417 | 57,070 | 1692 |
| P11 MglDKe04 | 1 | MiSeq | 2,243,640 | 1,160,159 | 19,312 | 413 |
| P12 MglDKe05 | 1 | MiSeq | 2,702,868 | 1,374,479 | 12,927 | 247 |
| P7 MglLTU01 | 1 | MiSeq | 2,676,336 | 1,719,083 | 152,509 | 3004 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiltbrunner, M.; Heckel, G. Assessing Genome-Wide Diversity in European Hantaviruses through Sequence Capture from Natural Host Samples. Viruses 2020, 12, 749. https://doi.org/10.3390/v12070749
Hiltbrunner M, Heckel G. Assessing Genome-Wide Diversity in European Hantaviruses through Sequence Capture from Natural Host Samples. Viruses. 2020; 12(7):749. https://doi.org/10.3390/v12070749
Chicago/Turabian StyleHiltbrunner, Melanie, and Gerald Heckel. 2020. "Assessing Genome-Wide Diversity in European Hantaviruses through Sequence Capture from Natural Host Samples" Viruses 12, no. 7: 749. https://doi.org/10.3390/v12070749
APA StyleHiltbrunner, M., & Heckel, G. (2020). Assessing Genome-Wide Diversity in European Hantaviruses through Sequence Capture from Natural Host Samples. Viruses, 12(7), 749. https://doi.org/10.3390/v12070749