Histone Deacetylase 6 Knockout Mice Exhibit Higher Susceptibility to Influenza A Virus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Infection
2.3. Quantitative Real-Time PCR
2.4. Western Blotting
2.5. Statistical Analysis
3. Results
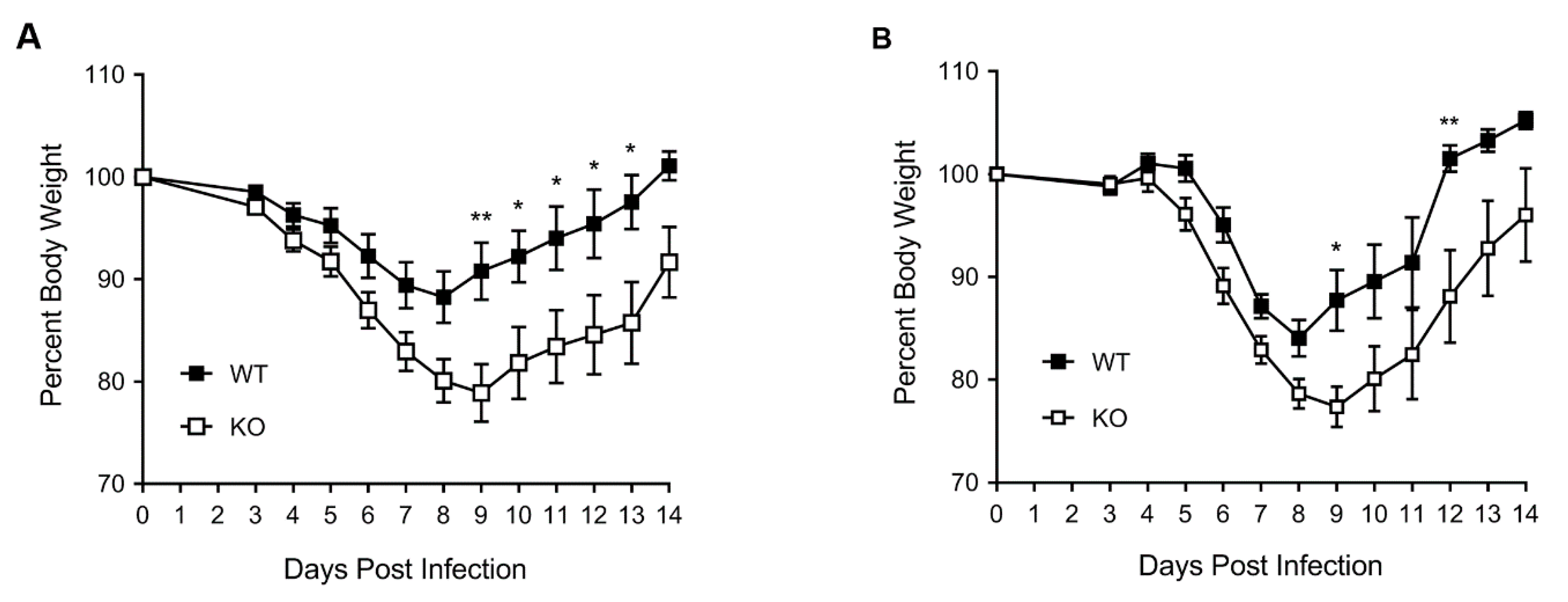
3.1. HDAC6 KO Mice Show Higher Weight Loss, Lower Survival and Higher Lung Virus Titres after IAV Infection
3.2. The Expression of Type I Interferon and Innate Antiviral Response Genes CD80, CXCL10, and IL15 Is Downregulated in HDAC6 KO Mouse Lungs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xue, K.S.; Bloom, J.D. Linking influenza virus evolution within and between human hosts. Virus Evol. 2020, 6, veaa010. [Google Scholar] [CrossRef]
- Sautto, G.A.; Kirchenbaum, G.A.; Ross, T.M. Towards a universal influenza vaccine: Different approaches for one goal. Virol. J. 2018, 15, 17. [Google Scholar] [CrossRef]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza virus: Dealing with a drifting and shifting pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- Liang, T.; Fang, H. Structure, functions and selective inhibitors of HDAC6. Curr. Top. Med. Chem. 2018, 18, 2429–2447. [Google Scholar] [CrossRef]
- Osko, J.D.; Christianson, D.W. Structural determinants of affinity and selectivity in the binding of inhibitors to histone deacetylase 6. Bioorg. Med. Chem. Lett. 2020, 30, 127023. [Google Scholar] [CrossRef]
- Husain, M.; Cheung, C.Y. Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J. Virol. 2014, 88, 11229–11239. [Google Scholar] [CrossRef]
- Nutsford, A.N.; Galvin, H.D.; Ahmed, F.; Husain, M. The Class IV human deacetylase, HDAC11, exhibits anti-influenza A virus properties via its involvement in host innate antiviral response. Cell Microbiol. 2019, 21, e12989. [Google Scholar] [CrossRef]
- Galvin, H.D.; Husain, M. Influenza A virus-induced host caspase and viral PA-X antagonize the antiviral host factor, histone deacetylase 4. J. Biol. Chem. 2019, 294, 20207–20221. [Google Scholar] [CrossRef]
- Nagesh, P.T.; Hussain, M.; Galvin, H.D.; Husain, M. Histone deacetylase 2 is a component of influenza A virus-induced host antiviral response. Front. Microbiol. 2017, 8, 1315. [Google Scholar] [CrossRef]
- Nagesh, P.T.; Husain, M. Influenza A virus dysregulates host histone deacetylase 1 that inhibits viral infection in lung epithelial cells. J. Virol. 2016, 90, 4614–4625. [Google Scholar] [CrossRef]
- Koyuncu, E.; Budayeva, H.G.; Miteva, Y.V.; Ricci, D.P.; Silhavy, T.J.; Shenk, T.; Cristea, I.M. Sirtuins are evolutionarily conserved viral restriction factors. mBio 2014, 5. [Google Scholar] [CrossRef]
- Choi, S.J.; Lee, H.C.; Kim, J.H.; Park, S.Y.; Kim, T.H.; Lee, W.K.; Jang, D.J.; Yoon, J.E.; Choi, Y.I.; Kim, S.; et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 2016, 35, 429–442. [Google Scholar] [CrossRef]
- Chen, H.; Qian, Y.; Chen, X.; Ruan, Z.; Ye, Y.; Chen, H.; Babiuk, L.A.; Jung, Y.S.; Dai, J. HDAC6 restricts influenza A virus by deacetylation of the RNA Polymerase PA subunit. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Gao, Y.S.; Hubbert, C.C.; Lu, J.; Lee, Y.S.; Lee, J.Y.; Yao, T.P. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell Biol. 2007, 27, 8637–8647. [Google Scholar] [CrossRef]
- Mahlknecht, U.; Schnittger, S.; Landgraf, F.; Schoch, C.; Ottmann, O.G.; Hiddemann, W.; Hoelzer, D. Assignment of the human histone deacetylase 6 gene (HDAC6) to X chromosome p11.23 by in situ hybridization. Cytogenet. Cell Genet. 2001, 93, 135–136. [Google Scholar] [CrossRef]
- Means, G.D.; Toy, D.Y.; Baum, P.R.; Derry, J.M.J. A transcript map of a 2-Mb BAC Contig in the proximal portion of the mouse X chromosome and regional mapping of the scurfy mutation. Genomics 2000, 65, 213–223. [Google Scholar] [CrossRef]
- Oslund, K.L.; Baumgarth, N. Influenza-induced innate immunity: Regulators of viral replication, respiratory tract pathology & adaptive immunity. Future Virol. 2011, 6, 951–962. [Google Scholar] [CrossRef]
- Husain, M.; Harrod, K.S. Influenza A virus-induced caspase-3 cleaves the histone deacetylase 6 in infected epithelial cells. FEBS Lett. 2009, 583, 2517–2520. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Harrod, K.S. Enhanced acetylation of alpha-tubulin in influenza A virus infected epithelial cells. FEBS Lett. 2011, 585, 128–132. [Google Scholar] [CrossRef]
- Liu, H.M.; Jiang, F.; Loo, Y.M.; Hsu, S.; Hsiang, T.Y.; Marcotrigiano, J.; Gale, M., Jr. Regulation of retinoic acid inducible Gene-I (RIG-I) activation by the histone deacetylase 6. EBioMedicine 2016, 9, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Meng, Q.; Huo, L.; Yang, M.; Wang, L.; Chen, X.; Wang, J.; Li, Z.; Ye, X.; Liu, N.; et al. Overexpression of Hdac6 enhances resistance to virus infection in embryonic stem cells and in mice. Protein Cell 2015, 6, 152–156. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A virus uses the aggresome processing machinery for host cell entry. Science (N. Y.) 2014, 346, 473–477. [Google Scholar] [CrossRef]
- Lu, X.; Masic, A.; Liu, Q.; Zhou, Y. Regulation of influenza A virus induced CXCL-10 gene expression requires PI3K/Akt pathway and IRF3 transcription factor. Mol. Immunol. 2011, 48, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Wagstaffe, H.R.; Nielsen, C.M.; Riley, E.M.; Goodier, M.R. IL-15 promotes polyfunctional NK cell responses to influenza by boosting IL-12 production. J. Immunol. 2018, 200, 2738–2747. [Google Scholar] [CrossRef]
- Fuse, S.; Obar, J.J.; Bellfy, S.; Leung, E.K.; Zhang, W.; Usherwood, E.J. CD80 and CD86 control antiviral CD8+ T-cell function and immune surveillance of murine gammaherpesvirus 68. J. Virol. 2006, 80, 9159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Coyne, C.B.; Sarkar, S.N. PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and β-catenin. EMBO J. 2011, 30, 4838–4849. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanin, M.; DeBeauchamp, J.; Vangala, G.; Webby, R.J.; Husain, M. Histone Deacetylase 6 Knockout Mice Exhibit Higher Susceptibility to Influenza A Virus Infection. Viruses 2020, 12, 728. https://doi.org/10.3390/v12070728
Zanin M, DeBeauchamp J, Vangala G, Webby RJ, Husain M. Histone Deacetylase 6 Knockout Mice Exhibit Higher Susceptibility to Influenza A Virus Infection. Viruses. 2020; 12(7):728. https://doi.org/10.3390/v12070728
Chicago/Turabian StyleZanin, Mark, Jennifer DeBeauchamp, Gowthami Vangala, Richard J. Webby, and Matloob Husain. 2020. "Histone Deacetylase 6 Knockout Mice Exhibit Higher Susceptibility to Influenza A Virus Infection" Viruses 12, no. 7: 728. https://doi.org/10.3390/v12070728
APA StyleZanin, M., DeBeauchamp, J., Vangala, G., Webby, R. J., & Husain, M. (2020). Histone Deacetylase 6 Knockout Mice Exhibit Higher Susceptibility to Influenza A Virus Infection. Viruses, 12(7), 728. https://doi.org/10.3390/v12070728