Viral Membrane Fusion and the Transmembrane Domain

{kind=link}
{kind=link}
Abstract
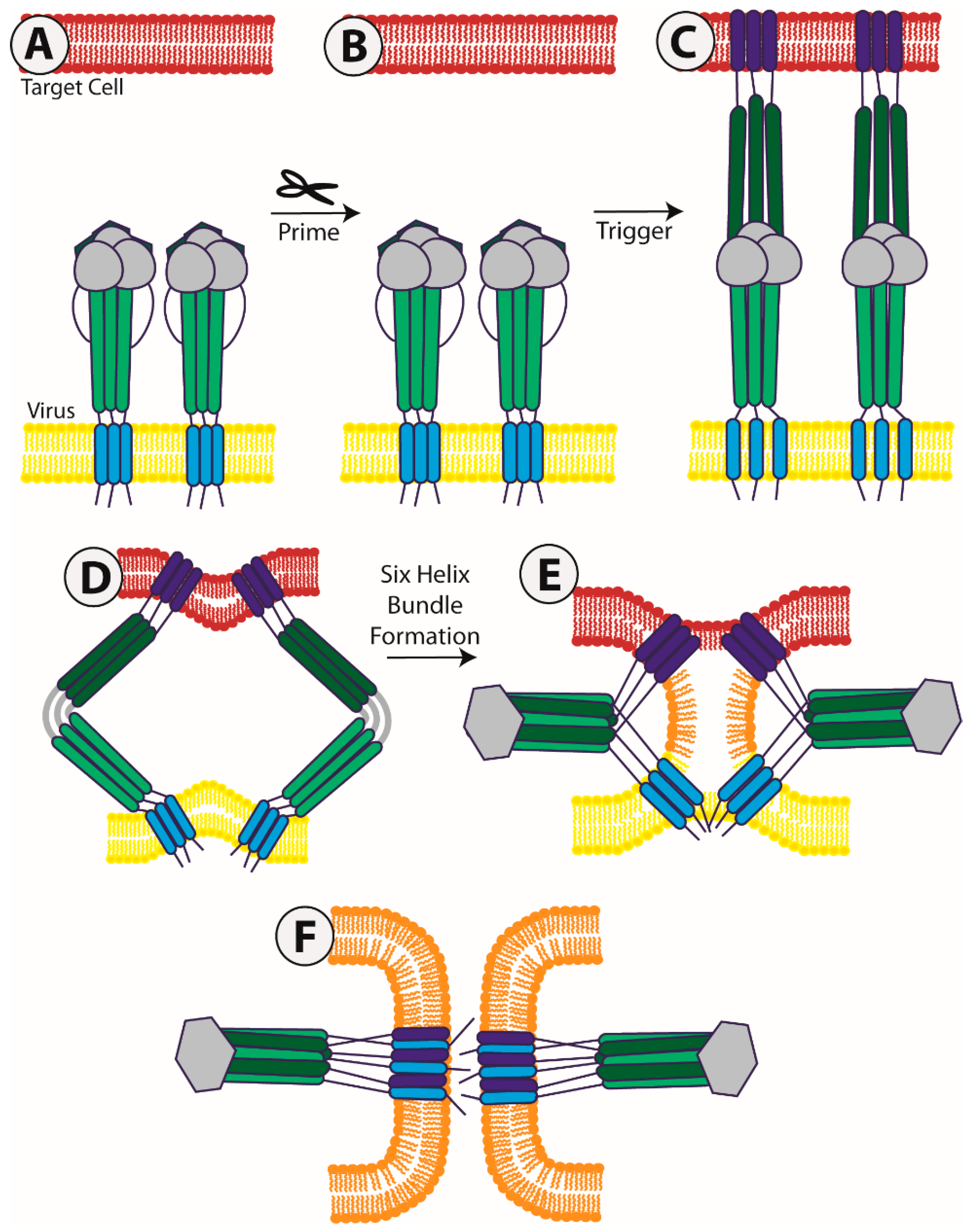
1. Introduction
2. Class I Fusion Proteins
2.1. Influenza
2.2. Human Immunodeficiency Virus (HIV)
2.3. Paramyxoviruses
2.4. Other Class I Viral Fusion Proteins
3. Class II Fusion Proteins
4. Class III Fusion Proteins
5. Targeting the TMD
6. Conclusions and Questions
Author Contributions
Funding
Conflicts of Interest
References
- Earp, L.J.; Delos, S.E.; Park, H.E.; White, J.M. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 2005, 285, 25–66. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef]
- Dutch, R.E.; Jardetzky, T.S.; Lamb, R.A. Virus membrane fusion proteins: Biological machines that undergo a metamorphosis. Biosci. Rep. 2000, 20, 597–612. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, P.I.; Zimmerberg, J.; Chizmadzhev, Y.A.; Cohen, F.S. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. USA 2001, 98, 7235–7240. [Google Scholar] [CrossRef] [PubMed]
- Parsegian, V.A.; Fuller, N.; Rand, R.P. Measured work of deformation and repulsion of lecithin bilayers. Proc. Natl. Acad. Sci. USA 1979, 76, 2750–2754. [Google Scholar] [CrossRef]
- Rand, R.P.; Parsegian, V.A. Physical force considerations in model and biological membranes. Can. J. Biochem. Cell Biol. 1984, 62, 752–759. [Google Scholar] [CrossRef]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef]
- Battles, M.B.; Mas, V.; Olmedillas, E.; Cano, O.; Vazquez, M.; Rodriguez, L.; Melero, J.A.; McLellan, J.S. Structure and immunogenicity of pre-fusion-stabilized human metapneumovirus F glycoprotein. Nat. Commun. 2017, 8, 1528. [Google Scholar] [CrossRef]
- Bizebard, T.; Gigant, B.; Rigolet, P.; Rasmussen, B.; Diat, O.; Bosecke, P.; Wharton, S.A.; Skehel, J.J.; Knossow, M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 1995, 376, 92–94. [Google Scholar] [CrossRef]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P.; Cupo, A.; Sok, D.; Stanfield, R.L.; Lyumkis, D.; Deller, M.C.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Cottrell, C.A.; Wang, N.; Pallesen, J.; Yassine, H.M.; Turner, H.L.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Pre-fusion structure of a human coronavirus spike protein. Nature 2016, 531, 118–121. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar] [CrossRef]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315, 843–848. [Google Scholar] [CrossRef]
- Stampfer, S.D.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 2010, 84, 12924–12933. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure and stabilization of the Hendra virus F glycoprotein in its prefusion form. Proc. Natl. Acad. Sci. USA 2016, 113, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chan, Y.P.; Bradel-Tretheway, B.; Akyol-Ataman, Z.; Zhu, Y.; Dutta, S.; Yan, L.; Feng, Y.; Wang, L.F.; Skiniotis, G.; et al. Crystal Structure of the Pre-fusion Nipah Virus Fusion Glycoprotein Reveals a Novel Hexamer-of-Trimers Assembly. PLoS Pathog. 2015, 11, e1005322. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Mothes, W. Structure and Dynamics of the Native HIV-1 Env Trimer. J. Virol. 2015, 89, 5752–5755. [Google Scholar] [CrossRef]
- Das, D.K.; Govindan, R.; Nikic-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 2018, 174, 926–937.e912. [Google Scholar] [CrossRef]
- Benhaim, M.; Lee, K.K. Single-Molecule Analysis of a Viral Fusion Protein Illuminates a Fusion-Active Intermediate State. Cell 2018, 174, 775–777. [Google Scholar] [CrossRef]
- Lai, A.L.; Freed, J.H. The Interaction between Influenza HA Fusion Peptide and Transmembrane Domain Affects Membrane Structure. Biophys. J. 2015, 109, 2523–2536. [Google Scholar] [CrossRef]
- Dobrowsky, T.M.; Zhou, Y.; Sun, S.X.; Siliciano, R.F.; Wirtz, D. Monitoring early fusion dynamics of human immunodeficiency virus type 1 at single-molecule resolution. J. Virol. 2008, 82, 7022–7033. [Google Scholar] [CrossRef]
- Langosch, D.; Hofmann, M.; Ungermann, C. The role of transmembrane domains in membrane fusion. Cell Mol. Life. Sci. 2007, 64, 850–864. [Google Scholar] [CrossRef]
- Kemble, G.W.; Danieli, T.; White, J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 1994, 76, 383–391. [Google Scholar] [CrossRef]
- Melikyan, G.B.; White, J.M.; Cohen, F.S. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J. Cell Biol. 1995, 131, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Compans, R.W. Alternative mechanisms of interaction between homotypic and heterotypic parainfluenza virus HN and F proteins. J. Gen. Virol. 1999, 80 Pt 1, 107–115. [Google Scholar] [CrossRef]
- Nussler, F.; Clague, M.J.; Herrmann, A. Meta-stability of the hemifusion intermediate induced by glycosylphosphatidylinositol-anchored influenza hemagglutinin. Biophys. J. 1997, 73, 2280–2291. [Google Scholar] [CrossRef][Green Version]
- Armstrong, R.T.; Kushnir, A.S.; White, J.M. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 2000, 151, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Roth, M.G.; Hunter, E. A chimeric avian retrovirus containing the influenza virus hemagglutinin gene has an expanded host range. J. Virol. 1992, 66, 7374–7382. [Google Scholar] [CrossRef]
- Melikyan, G.B.; Lin, S.; Roth, M.G.; Cohen, F.S. Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol. Biol. Cell 1999, 10, 1821–1836. [Google Scholar] [CrossRef][Green Version]
- Roth, M.G.; Doyle, C.; Sambrook, J.; Gething, M.J. Heterologous transmembrane and cytoplasmic domains direct functional chimeric influenza virus hemagglutinins into the endocytic pathway. J. Cell. Biol. 1986, 102, 1271–1283. [Google Scholar] [CrossRef]
- Schroth-Diez, B.; Ponimaskin, E.; Reverey, H.; Schmidt, M.F.; Herrmann, A. Fusion activity of transmembrane and cytoplasmic domain chimeras of the influenza virus glycoprotein hemagglutinin. J. Virol. 1998, 72, 133–141. [Google Scholar] [CrossRef]
- Taylor, G.M.; Sanders, D.A. The role of the membrane-spanning domain sequence in glycoprotein-mediated membrane fusion. Mol. Biol. Cell 1999, 10, 2803–2815. [Google Scholar] [CrossRef][Green Version]
- Langosch, D.; Heringa, J. Interaction of transmembrane helices by a knobs-into-holes packing characteristic of soluble coiled coils. Proteins 1998, 31, 150–159. [Google Scholar] [CrossRef]
- Markosyan, R.M.; Cohen, F.S.; Melikyan, G.B. The lipid-anchored ectodomai of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol. Biol. Cell 2000, 11, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Chen, J.; Skehel, J.J.; Wiley, D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA 1999, 96, 8967–8972. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Sabesan, S.; Skehel, J.J.; Wiley, D.C. Binding of the influenza A virus to cell-surface receptors: Structures of five hemagglutinin-sialyloligosaccharide complexes determined by X-ray crystallography. Virology 1997, 232, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 2004, 303, 1838–1842. [Google Scholar] [CrossRef]
- Sauter, N.K.; Glick, G.D.; Crowther, R.L.; Park, S.J.; Eisen, M.B.; Skehel, J.J.; Knowles, J.R.; Wiley, D.C. Crystallographic detection of a second ligand binding site in influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA 1992, 89, 324–328. [Google Scholar] [CrossRef]
- Stevens, J.; Corper, A.L.; Basler, C.F.; Taubenberger, J.K.; Palese, P.; Wilson, I.A. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 2004, 303, 1866–1870. [Google Scholar] [CrossRef]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Benton, D.J.; Nans, A.; Calder, L.J.; Turner, J.; Neu, U.; Lin, Y.P.; Ketelaars, E.; Kallewaard, N.L.; Corti, D.; Lanzavecchia, A.; et al. Influenza hemagglutinin membrane anchor. Proc. Natl. Acad. Sci. USA 2018, 115, 10112–10117. [Google Scholar] [CrossRef] [PubMed]
- Victor, B.L.; Baptista, A.M.; Soares, C.M. Structural determinants for the membrane insertion of the transmembrane peptide of hemagglutinin from influenza virus. J. Chem. Inf. Modeling 2012, 52, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ge, P.; Yu, X.; Brannan, J.M.; Bi, G.; Zhang, Q.; Schein, S.; Zhou, Z.H. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat. Struct. Mol. Biol. 2013, 20, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.S.; Georgieva, E.R.; Borbat, P.P.; Freed, J.H.; Heldwein, E.E. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat. Struct. Mol. Biol. 2018, 25, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Gamblin, S.J.; Rosenthal, P.B.; Skehel, J.J. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Calder, L.J.; Rosenthal, P.B. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat. Struct. Mol. Biol. 2016, 23, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Cheng, S.F.; Kantchev, E.A.; Lin, C.H.; Liu, Y.T. Membrane interaction and structure of the transmembrane domain of influenza hemagglutinin and its fusion peptide complex. BMC Biol. 2008, 6, 2. [Google Scholar] [CrossRef]
- Ge, M.; Freed, J.H. Two conserved residues are important for inducing highly ordered membrane domains by the transmembrane domain of influenza hemagglutinin. Biophys. J. 2011, 100, 90–97. [Google Scholar] [CrossRef]
- Qiao, H.; Armstrong, R.T.; Melikyan, G.B.; Cohen, F.S.; White, J.M. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol. Biol. Cell 1999, 10, 2759–2769. [Google Scholar] [CrossRef]
- Ranaweera, A.; Ratnayake, P.U.; Ekanayaka, E.A.P.; Declercq, R.; Weliky, D.P. Hydrogen-Deuterium Exchange Supports Independent Membrane-Interfacial Fusion Peptide and Transmembrane Domains in Subunit 2 of Influenza Virus Hemagglutinin Protein, a Structured and Aqueous-Protected Connection between the Fusion Peptide and Soluble Ectodomain, and the Importance of Membrane Apposition by the Trimer-of-Hairpins Structure. Biochemistry 2019, 58, 2432–2446. [Google Scholar] [CrossRef]
- Engel, S.; de Vries, M.; Herrmann, A.; Veit, M. Mutation of a raft-targeting signal in the transmembrane region retards transport of influenza virus hemagglutinin through the Golgi. FEBS Lett. 2012, 586, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Hofer, C.T.; Thiele, C.; Veit, M. Cholesterol Binding to the Transmembrane Region of a Group 2 Hemagglutinin (HA) of Influenza Virus Is Essential for Virus Replication, Affecting both Virus Assembly and HA Fusion Activity. J. Virol. 2019, 93, e00555-19. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.L.; Frisz, J.F.; Klitzing, H.A.; Zimmerberg, J.; Weber, P.K.; Kraft, M.L. Hemagglutinin clusters in the plasma membrane are not enriched with cholesterol and sphingolipids. Biophys. J. 2015, 108, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Kordyukova, L.V.; Serebryakova, M.V.; Baratova, L.A.; Veit, M. S acylation of the hemagglutinin of influenza viruses: Mass spectrometry reveals site-specific attachment of stearic acid to a transmembrane cysteine. J. Virol. 2008, 82, 9288–9292. [Google Scholar] [CrossRef]
- Scheiffele, P.; Roth, M.G.; Simons, K. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997, 16, 5501–5508. [Google Scholar] [CrossRef]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar] [CrossRef]
- Veit, M.; Kretzschmar, E.; Kuroda, K.; Garten, W.; Schmidt, M.F.; Klenk, H.D.; Rott, R. Site-specific mutagenesis identifies three cysteine residues in the cytoplasmic tail as acylation sites of influenza virus hemagglutinin. J. Virol. 1991, 65, 2491–2500. [Google Scholar] [CrossRef]
- Wagner, R.; Herwig, A.; Azzouz, N.; Klenk, H.D. Acylation-mediated membrane anchoring of avian influenza virus hemagglutinin is essential for fusion pore formation and virus infectivity. J. Virol. 2005, 79, 6449–6458. [Google Scholar] [CrossRef]
- Barman, S.; Nayak, D.P. Analysis of the transmembrane domain of influenza virus neuraminidase, a type II transmembrane glycoprotein, for apical sorting and raft association. J. Virol. 2000, 74, 6538–6545. [Google Scholar] [CrossRef]
- Shang, L.; Yue, L.; Hunter, E. Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J. Virol. 2008, 82, 5417–5428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, B.; Chou, J.J. Structure of the transmembrane domain of HIV-1 envelope glycoprotein. FEBS J. 2017, 284, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Dev, J.; Park, D.; Fu, Q.; Chen, J.; Ha, H.J.; Ghantous, F.; Herrmann, T.; Chang, W.; Liu, Z.; Frey, G.; et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science 2016, 353, 172–175. [Google Scholar] [CrossRef]
- Gangupomu, V.K.; Abrams, C.F. All-atom models of the membrane-spanning domain of HIV-1 gp41 from metadynamics. Biophys. J. 2010, 99, 3438–3444. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Shaik, M.M.; Cai, Y.; Ghantous, F.; Piai, A.; Peng, H.; Rits-Volloch, S.; Liu, Z.; Harrison, S.C.; Seaman, M.S.; et al. Structure of the membrane proximal external region of HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2018, 115, E8892–E8899. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.; Lee, M.; Waring, A.J.; Hong, M. Oligomeric Structure and Three-Dimensional Fold of the HIV gp41 Membrane-Proximal External Region and Transmembrane Domain in Phospholipid Bilayers. J. Am. Chem. Soc. 2018, 140, 8246–8259. [Google Scholar] [CrossRef] [PubMed]
- Apellaniz, B.; Rujas, E.; Serrano, S.; Morante, K.; Tsumoto, K.; Caaveiro, J.M.; Jimenez, M.A.; Nieva, J.L. The Atomic Structure of the HIV-1 gp41 Transmembrane Domain and Its Connection to the Immunogenic Membrane-proximal External Region. J. Biol. Chem. 2015, 290, 12999–13015. [Google Scholar] [CrossRef] [PubMed]
- Buzon, V.; Natrajan, G.; Schibli, D.; Campelo, F.; Kozlov, M.M.; Weissenhorn, W. Crystal Structure of HIV-1 gp41 Including Both Fusion Peptide and Membrane Proximal External Regions. PLoS Pathog. 2010, 6, e1000880. [Google Scholar] [CrossRef]
- Lee, J.H.; Ozorowski, G.; Ward, A.B. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 2016, 351, 1043–1048. [Google Scholar] [CrossRef]
- Reardon, P.N.; Sage, H.; Dennison, S.M.; Martin, J.W.; Donald, B.R.; Alam, S.M.; Haynes, B.F.; Spicer, L.D. Structure of an HIV-1-neutralizing antibody target, the lipid-bound gp41 envelope membrane proximal region trimer. Proc. Natl. Acad. Sci. USA 2014, 111, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, G.; Briggs, J.A.; Grunewald, K.; Sattentau, Q.J.; Fuller, S.D. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2006, 2, e83. [Google Scholar] [CrossRef] [PubMed]
- Reuven, E.M.; Dadon, Y.; Viard, M.; Manukovsky, N.; Blumenthal, R.; Shai, Y. HIV-1 gp41 transmembrane domain interacts with the fusion peptide: Implication in lipid mixing and inhibition of virus-cell fusion. Biochemistry 2012, 51, 2867–2878. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Morgan, C.A.; Hong, M. Fully hydrophobic HIV gp41 adopts a hemifusion-like conformation in phospholipid bilayers. J. Biol. Chem. 2019, 294, 14732–14744. [Google Scholar] [CrossRef]
- Kondo, N.; Miyauchi, K.; Meng, F.; Iwamoto, A.; Matsuda, Z. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 2010, 285, 14681–14688. [Google Scholar] [CrossRef]
- Long, Y.; Meng, F.; Kondo, N.; Iwamoto, A.; Matsuda, Z. Conserved arginine residue in the membrane-spanning domain of HIV-1 gp41 is required for efficient membrane fusion. Protein Cell 2011, 2, 369–376. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hollingsworth, L.R.t.; Lemkul, J.A.; Bevan, D.R.; Brown, A.M. HIV-1 Env gp41 Transmembrane Domain Dynamics Are Modulated by Lipid, Water, and Ion Interactions. Biophys. J. 2018, 115, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.K.; Gangupomu, V.K.; Abrams, C.F. Characterization of the water defect at the HIV-1 gp41 membrane spanning domain in bilayers with and without cholesterol using molecular simulations. Biochim. Biophys. Acta 2014, 1838, 1396–1405. [Google Scholar] [CrossRef]
- Piai, A.; Dev, J.; Fu, Q.; Chou, J.J. Stability and Water Accessibility of the Trimeric Membrane Anchors of the HIV-1 Envelope Spikes. J. Am. Chem. Soc. 2017, 139, 18432–18435. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Faingold, O.; Kaushansky, N.; Ben-Nun, A.; Shai, Y. A highly conserved sequence associated with the HIV gp41 loop region is an immunomodulator of antigen-specific T cells in mice. Blood 2013, 121, 2244–2252. [Google Scholar] [CrossRef]
- Bloch, I.; Quintana, F.J.; Gerber, D.; Cohen, T.; Cohen, I.R.; Shai, Y. T-cell inactivation and immunosuppressive activity induced by HIV gp41 via novel interacting motif. FASEB J. 2007, 21, 393–401. [Google Scholar] [CrossRef]
- Rotem, E.; Reuven, E.M.; Klug, Y.A.; Shai, Y. The Transmembrane Domain of HIV-1 gp41 Inhibits T-Cell Activation by Targeting Multiple T-Cell Receptor Complex Components through Its GxxxG Motif. Biochemistry 2016, 55, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Klug, Y.A.; Rotem, E.; Schwarzer, R.; Shai, Y. Mapping out the intricate relationship of the HIV envelope protein and the membrane environment. Biochim. Biophys. Acta Biomembr. 2017, 1859, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Reuven, E.M.; Ali, M.; Rotem, E.; Schwarzer, R.; Gramatica, A.; Futerman, A.H.; Shai, Y. The HIV-1 envelope transmembrane domain binds TLR2 through a distinct dimerization motif and inhibits TLR2-mediated responses. PLoS Pathog. 2014, 10, e1004248. [Google Scholar] [CrossRef]
- Faingold, O.; Cohen, T.; Shai, Y. A GxxxG-like motif within HIV-1 fusion peptide is critical to its immunosuppressant activity, structure, and interaction with the transmembrane domain of the T-cell receptor. J. Biol. Chem. 2012, 287, 33503–33511. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Pevsner-Fischer, M.; Cohen, N.; Cohen, I.R.; Shai, Y. Characterization of the interacting domain of the HIV-1 fusion peptide with the transmembrane domain of the T-cell receptor. Biochemistry 2008, 47, 4826–4833. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Gerber, D.; Kent, S.C.; Cohen, I.R.; Shai, Y. HIV-1 fusion peptide targets the TCR and inhibits antigen-specific T cell activation. J. Clin. Invest. 2005, 115, 2149–2158. [Google Scholar] [CrossRef]
- Reichart, T.M.; Baksh, M.M.; Rhee, J.K.; Fiedler, J.D.; Sligar, S.G.; Finn, M.G.; Zwick, M.B.; Dawson, P.E. Trimerization of the HIV Transmembrane Domain in Lipid Bilayers Modulates Broadly Neutralizing Antibody Binding. Angew. Chem. Int. Ed. Engl. 2016, 55, 2688–2692. [Google Scholar] [CrossRef]
- Torrents de la Pena, A.; Rantalainen, K.; Cottrell, C.A.; Allen, J.D.; van Gils, M.J.; Torres, J.L.; Crispin, M.; Sanders, R.W.; Ward, A.B. Similarities and differences between native HIV-1 envelope glycoprotein trimers and stabilized soluble trimer mimetics. PLoS Pathog. 2019, 15, e1007920. [Google Scholar] [CrossRef]
- Wang, Y.; Kaur, P.; Sun, Z.J.; Elbahnasawy, M.A.; Hayati, Z.; Qiao, Z.S.; Bui, N.N.; Chile, C.; Nasr, M.L.; Wagner, G.; et al. Topological analysis of the gp41 MPER on lipid bilayers relevant to the metastable HIV-1 envelope prefusion state. Proc. Natl. Acad. Sci. USA 2019, 116, 22556–22566. [Google Scholar] [CrossRef]
- Pinto, D.; Fenwick, C.; Caillat, C.; Silacci, C.; Guseva, S.; Dehez, F.; Chipot, C.; Barbieri, S.; Minola, A.; Jarrossay, D.; et al. Structural Basis for Broad HIV-1 Neutralization by the MPER-Specific Human Broadly Neutralizing Antibody LN01. Cell Host Microbe 2019, 26, 623–637.e628. [Google Scholar] [CrossRef]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef]
- Miyauchi, K.; Curran, A.R.; Long, Y.; Kondo, N.; Iwamoto, A.; Engelman, D.M.; Matsuda, Z. The membrane-spanning domain of gp41 plays a critical role in intracellular trafficking of the HIV envelope protein. Retrovirology 2010, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Perrin, J.; Bary, A.; Vernay, A.; Cosson, P. Role of the HIV-1 envelope transmembrane domain in intracellular sorting. BMC Cell Biol. 2018, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Yao, H.; Kwon, B.; Waring, A.J.; Ruchala, P.; Singh, C.; Hong, M. Conformation and Trimer Association of the Transmembrane Domain of the Parainfluenza Virus Fusion Protein in Lipid Bilayers from Solid-State NMR: Insights into the Sequence Determinants of Trimer Structure and Fusion Activity. J. Mol. Biol. 2018, 430, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Lee, M.W.; Waring, A.J.; Wong, G.C.; Hong, M. Viral fusion protein transmembrane domain adopts beta-strand structure to facilitate membrane topological changes for virus-cell fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 10926–10931. [Google Scholar] [CrossRef]
- Bissonnette, M.L.; Donald, J.E.; DeGrado, W.F.; Jardetzky, T.S.; Lamb, R.A. Functional analysis of the transmembrane domain in paramyxovirus F protein-mediated membrane fusion. J. Mol. Biol. 2009, 386, 14–36. [Google Scholar] [CrossRef]
- Afonso, C.L.; Amarasinghe, G.K.; Banyai, K.; Bao, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.X.; Briese, T.; et al. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef]
- Smith, E.C.; Smith, S.E.; Carter, J.R.; Webb, S.R.; Gibson, K.M.; Hellman, L.M.; Fried, M.G.; Dutch, R.E. Trimeric transmembrane domain interactions in paramyxovirus fusion proteins: Roles in protein folding, stability, and function. J. Biol. Chem. 2013, 288, 35726–35735. [Google Scholar] [CrossRef]
- Webb, S.; Nagy, T.; Moseley, H.; Fried, M.; Dutch, R. Hendra virus fusion protein transmembrane domain contributes to pre-fusion protein stability. J. Biol. Chem. 2017, 292, 5685–5694. [Google Scholar] [CrossRef]
- Gravel, K.A.; McGinnes, L.W.; Reitter, J.; Morrison, T.G. The transmembrane domain sequence affects the structure and function of the Newcastle disease virus fusion protein. J. Virol. 2011, 85, 3486–3497. [Google Scholar] [CrossRef]
- Branttie, J.M.; Dutch, R.E. Parainfluenza virus 5 fusion protein maintains pre-fusion stability but not fusogenic activity following mutation of a transmembrane leucine/isoleucine domain. J. Gen. Virol. 2020, 101, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, K.B.; Dutch, R.E. Transmembrane Domain Dissociation Is Required for Hendra Virus F Protein Fusogenic Activity. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Culler, M.R.; Hellman, L.M.; Fried, M.G.; Creamer, T.P.; Dutch, R.E. Beyond anchoring: The expanding role of the hendra virus fusion protein transmembrane domain in protein folding, stability, and function. J. Virol. 2012, 86, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Zokarkar, A.; Connolly, S.A.; Jardetzky, T.S.; Lamb, R.A. Reversible Inhibition of Fusion Activity of a Paramyxovirus Fusion Protein by an Engineered Disulfide Bond in the Membrane-Proximal External Region. J. Virol. 2012, 86, 12397–12401. [Google Scholar] [CrossRef][Green Version]
- Eckert, D.M.; Kim, P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001, 70, 777–810. [Google Scholar] [CrossRef]
- Plattet, P.; Plemper, R.K. Envelope protein dynamics in paramyxovirus entry. mBio 2013, 4, e00413. [Google Scholar] [CrossRef]
- Yao, H.; Lee, M.; Liao, S.Y.; Hong, M. Solid-State Nuclear Magnetic Resonance Investigation of the Structural Topology and Lipid Interactions of a Viral Fusion Protein Chimera Containing the Fusion Peptide and Transmembrane Domain. Biochemistry 2016, 55, 6787–6800. [Google Scholar] [CrossRef] [PubMed]
- Donald, J.E.; Zhang, Y.; Fiorin, G.; Carnevale, V.; Slochower, D.R.; Gai, F.; Klein, M.L.; DeGrado, W.F. Transmembrane orientation and possible role of the fusogenic peptide from parainfluenza virus 5 (PIV5) in promoting fusion. Proc. Natl. Acad. Sci. USA 2011, 108, 3958–3963. [Google Scholar] [CrossRef]
- Weise, K.; Reed, J. Fusion peptides and transmembrane domains of fusion proteins are characterized by different but specific structural properties. Chembiochem 2008, 9, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Munoz, N.; Sun, W.; Ray, G.; Schmitt, P.T.; Webb, S.; Gibson, K.; Dutch, R.E.; Schmitt, A.P. Mutations in the Transmembrane Domain and Cytoplasmic Tail of Hendra Virus Fusion Protein Disrupt Virus-Like-Particle Assembly. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Popa, A.; Carter, J.R.; Smith, S.E.; Hellman, L.; Fried, M.G.; Dutch, R.E. Residues in the hendra virus fusion protein transmembrane domain are critical for endocytic recycling. J. Virol. 2012, 86, 3014–3026. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.M.; Zu, S.L.; Zhou, W.W.; Wang, X.J.; Shuai, L.; Wang, X.L.; Ge, J.Y.; Bu, Z.G. Chimeric rabies glycoprotein with a transmembrane domain and cytoplasmic tail from Newcastle disease virus fusion protein incorporates into the Newcastle disease virion at reduced levels. J. Vet. Sci. 2017, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Pager, C.T.; Craft, W.W., Jr.; Patch, J.; Dutch, R.E. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 2006, 346, 251–257. [Google Scholar] [CrossRef]
- Pager, C.T.; Dutch, R.E. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 2005, 79, 12714–12720. [Google Scholar] [CrossRef] [PubMed]
- Pager, C.T.; Wurth, M.A.; Dutch, R.E. Subcellular localization and calcium and pH requirements for proteolytic processing of the Hendra virus fusion protein. J. Virol. 2004, 78, 9154–9163. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, L.; Aguilar, H.C.; Chou, K.C. A stochastic assembly model for Nipah virus revealed by super-resolution microscopy. Nat. Commun. 2018, 9, 3050. [Google Scholar] [CrossRef] [PubMed]
- Mattera, R.; Farias, G.G.; Mardones, G.A.; Bonifacino, J.S. Co-assembly of viral envelope glycoproteins regulates their polarized sorting in neurons. PLoS Pathog. 2014, 10, e1004107. [Google Scholar] [CrossRef]
- Webb, S.R.; Smith, S.E.; Fried, M.G.; Dutch, R.E. Transmembrane Domains of Highly Pathogenic Viral Fusion Proteins Exhibit Trimeric Association In Vitro. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Lee, J.; Nyenhuis, D.A.; Nelson, E.A.; Cafiso, D.S.; White, J.M.; Tamm, L.K. Structure of the Ebola virus envelope protein MPER/TM domain and its interaction with the fusion loop explains their fusion activity. Proc. Natl. Acad. Sci. USA 2017, 114, E7987–E7996. [Google Scholar] [CrossRef]
- Beniac, D.R.; Booth, T.F. Structure of the Ebola virus glycoprotein spike within the virion envelope at 11 A resolution. Sci. Rep. 2017, 7, 46374. [Google Scholar] [CrossRef]
- Liu, N.; Girvin, M.E.; Brenowitz, M.; Lai, J.R. Conformational and lipid bilayer-perturbing properties of Marburg virus GP2 segments containing the fusion loop and membrane-proximal external region/transmembrane domain. Heliyon 2019, 5, e03018. [Google Scholar] [CrossRef] [PubMed]
- Corver, J.; Broer, R.; van Kasteren, P.; Spaan, W. Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: Refinement of the requirements for SARS coronavirus cell entry. Virol. J. 2009, 6, 230. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Kataoka, M.; Shirato, K.; Matsuyama, S. Biochemical Analysis of Coronavirus Spike Glycoprotein Conformational Intermediates during Membrane Fusion. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.A.; Dutch, R.E.; Lamb, R.A.; Jardetzky, T.S. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell 1999, 3, 309–319. [Google Scholar] [CrossRef]
- Neil, S.J. The antiviral activities of tetherin. Curr. Top. Microbiol. Immunol. 2013, 371, 67–104. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Gnirss, K.; Fiedler, M.; Kramer-Kuhl, A.; Bolduan, S.; Mittler, E.; Becker, S.; Schindler, M.; Pohlmann, S. Analysis of determinants in filovirus glycoproteins required for tetherin antagonism. Viruses 2014, 6, 1654–1671. [Google Scholar] [CrossRef]
- Vande Burgt, N.H.; Kaletsky, R.L.; Bates, P. Requirements within the Ebola Viral Glycoprotein for Tetherin Antagonism. Viruses 2015, 7, 5587–5602. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, M.; Hoffmann, M.; Brinkmann, C.; Nehls, J.; Winkler, M.; Schindler, M.; Pohlmann, S. A GXXXA Motif in the Transmembrane Domain of the Ebola Virus Glycoprotein Is Required for Tetherin Antagonism. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Hacke, M.; Bjorkholm, P.; Hellwig, A.; Himmels, P.; Ruiz de Almodovar, C.; Brugger, B.; Wieland, F.; Ernst, A.M. Inhibition of Ebola virus glycoprotein-mediated cytotoxicity by targeting its transmembrane domain and cholesterol. Nat. Commun. 2015, 6, 7688. [Google Scholar] [CrossRef]
- Effantin, G.; Estrozi, L.F.; Aschman, N.; Renesto, P.; Stanke, N.; Lindemann, D.; Schoehn, G.; Weissenhorn, W. Cryo-electron Microscopy Structure of the Native Prototype Foamy Virus Glycoprotein and Virus Architecture. PLoS Pathog. 2016, 12, e1005721. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, H.; Liu, Q.M.; Sun, Y.; Li, Z.; Liu, W.H.; He, X.H.; Song, J.; Wang, Y.X. Structure of transmembrane subunits gp47 of the foamy virus envelope glycoproteins. Acta Virol. 2016, 60, 181–189. [Google Scholar] [CrossRef] [PubMed]
- York, J.; Romanowski, V.; Lu, M.; Nunberg, J.H. The signal peptide of the Junín arenavirus envelope glycoprotein is myristoylated and forms an essential subunit of the mature G1-G2 complex. J. Virol. 2004, 78, 10783–10792. [Google Scholar] [CrossRef] [PubMed]
- York, J.; Nunberg, J.H. Role of the stable signal peptide of Junín arenavirus envelope glycoprotein in pH-dependent membrane fusion. J. Virol. 2006, 80, 7775–7780. [Google Scholar] [CrossRef]
- Agnihothram, S.S.; York, J.; Trahey, M.; Nunberg, J.H. Bitopic membrane topology of the stable signal peptide in the tripartite Junín virus GP-C envelope glycoprotein complex. J. Virol. 2007, 81, 4331–4337. [Google Scholar] [CrossRef] [PubMed]
- Messina, E.L.; York, J.; Nunberg, J.H. Dissection of the role of the stable signal peptide of the arenavirus envelope glycoprotein in membrane fusion. J. Virol. 2012, 86, 6138–6145. [Google Scholar] [CrossRef]
- York, J.; Nunberg, J.H. Intersubunit interactions modulate pH-induced activation of membrane fusion by the Junin virus envelope glycoprotein GPC. J. Virol. 2009, 83, 4121–4126. [Google Scholar] [CrossRef]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Wengler, G.; Rey, F.A. The Fusion Glycoprotein Shell of Semliki Forest Virus. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef]
- Kielian, M. Class II virus membrane fusion proteins. Virology 2006, 344, 38–47. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Modis, Y. Structural models of the membrane anchors of envelope glycoproteins E1 and E2 from pestiviruses. Virology 2014, 454–455, 93–101. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 A resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hryc, C.F.; Cong, Y.; Liu, X.; Jakana, J.; Gorchakov, R.; Baker, M.L.; Weaver, S.C.; Chiu, W. 4.4 A cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 2011, 30, 3854–3863. [Google Scholar] [CrossRef]
- Fritz, R.; Blazevic, J.; Taucher, C.; Pangerl, K.; Heinz, F.X.; Stiasny, K. The unique transmembrane hairpin of flavivirus fusion protein E is essential for membrane fusion. J. Virol. 2011, 85, 4377–4385. [Google Scholar] [CrossRef] [PubMed]
- Ronecker, S.; Zimmer, G.; Herrler, G.; Greiser-Wilke, I.; Grummer, B. Formation of bovine viral diarrhea virus E1-E2 heterodimers is essential for virus entry and depends on charged residues in the transmembrane domains. J. Gen. Virol. 2008, 89, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.C.; Spear, P.G. Biochemistry. Viral glycoproteins and an evolutionary conundrum. Science 2006, 313, 177–178. [Google Scholar] [CrossRef]
- Backovic, M.; Jardetzky, T.S. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 2009, 19, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar] [CrossRef]
- Kadlec, J.; Loureiro, S.; Abrescia, N.G.; Stuart, D.I.; Jones, I.M. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008, 15, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Vallbracht, M.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Functional Relevance of the Transmembrane Domain and Cytoplasmic Tail of the Pseudorabies Virus Glycoprotein H for Membrane Fusion. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Dennison, S.M.; Greenfield, N.; Lenard, J.; Lentz, B.R. VSV Transmembrane Domain (TMD) Peptide Promotes PEG-Mediated Fusion of Liposomes in a Conformationally Sensitive Fashion†. Biochemistry 2002, 41, 14925–14934. [Google Scholar] [CrossRef]
- Odell, D.; Wanas, E.; Yan, J.; Ghosh, H.P. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of vesicular stomatitis virus glycoprotein G. J. Virol. 1997, 71, 7996–8000. [Google Scholar] [CrossRef]
- Cleverley, D.Z.; Lenard, J. The transmembrane domain in viral fusion: Essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. USA 1998, 95, 3425–3430. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, T.; Chakraborty, H.; Lentz, B.R. The transmembrane domain peptide of vesicular stomatitis virus promotes both intermediate and pore formation during PEG-mediated vesicle fusion. Biophys. J. 2014, 107, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.; Martinez-Gil, L.; Tamborero, S.; Mingarro, I. Membrane topology of gp41 and amyloid precursor protein: Interfering transmembrane interactions as potential targets for HIV and Alzheimer treatment. Biochim. Biophys. Acta 2009, 1788, 2132–2141. [Google Scholar] [CrossRef]
- Roth, L.; Nasarre, C.; Dirrig-Grosch, S.; Aunis, D.; Cremel, G.; Hubert, P.; Bagnard, D. Transmembrane domain interactions control biological functions of neuropilin-1. Mol. Biol. Cell 2008, 19, 646–654. [Google Scholar] [CrossRef]
- Arpel, A.; Sawma, P.; Spenle, C.; Fritz, J.; Meyer, L.; Garnier, N.; Velazquez-Quesada, I.; Hussenet, T.; Aci-Seche, S.; Baumlin, N.; et al. Transmembrane domain targeting peptide antagonizing ErbB2/Neu inhibits breast tumor growth and metastasis. Cell Rep. 2014, 8, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Z.; Xu, L.; Yang, H.; Liu, W. Transmembrane domain dependent inhibitory function of FcgammaRIIB. Protein Cell 2018, 9, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, W.; Ananthan, S.; Suto, M.J.; Li, Y. Discovery of novel frizzled-7 inhibitors by targeting the receptor’s transmembrane domain. Oncotarget 2017, 8, 91459–91470. [Google Scholar] [CrossRef]
- Pessi, A.; Langella, A.; Capito, E.; Ghezzi, S.; Vicenzi, E.; Poli, G.; Ketas, T.; Mathieu, C.; Cortese, R.; Horvat, B.; et al. A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS ONE 2012, 7, e36833. [Google Scholar] [CrossRef]
- Nasarre, C.; Roth, M.; Jacob, L.; Roth, L.; Koncina, E.; Thien, A.; Labourdette, G.; Poulet, P.; Hubert, P.; Cremel, G.; et al. Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene 2010, 29, 2381–2392. [Google Scholar] [CrossRef]
- Jacob, L.; Sawma, P.; Garnier, N.; Meyer, L.A.; Fritz, J.; Hussenet, T.; Spenle, C.; Goetz, J.; Vermot, J.; Fernandez, A.; et al. Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget 2016, 7, 57851–57865. [Google Scholar] [CrossRef] [PubMed]
- Goh, E.T.H.; Lin, Z.; Ahn, B.Y.; Lopes-Rodrigues, V.; Dang, N.H.; Salim, S.; Berger, B.; Dymock, B.; Senger, D.L.; Ibanez, C.F. A Small Molecule Targeting the Transmembrane Domain of Death Receptor p75(NTR) Induces Melanoma Cell Death and Reduces Tumor Growth. Cell Chem. Biol. 2018, 25, 1485–1494.e1485. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Saludes, J.P.; Zhao, T.X.; Csakai, A.; Fiorini, Z.; Chavez, S.A.; Li, J.; Lee, G.I.; Varga, K.; Yin, H. Targeting the lateral interactions of transmembrane domain 5 of Epstein-Barr virus latent membrane protein 1. Biochim. Biophys. Acta 2012, 1818, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.T.; Webb, S.R.; Dutch, R.E. A Hydrophobic Target: Using the Paramyxovirus Fusion Protein Transmembrane Domain To Modulate Fusion Protein Stability. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.G.; Yang, P.L.; Harrison, S.C. Peptide inhibitors of flavivirus entry derived from the E protein stem. J. Virol. 2010, 84, 12549–12554. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrett, C.T.; Dutch, R.E. Viral Membrane Fusion and the Transmembrane Domain. Viruses 2020, 12, 693. https://doi.org/10.3390/v12070693
Barrett CT, Dutch RE. Viral Membrane Fusion and the Transmembrane Domain. Viruses. 2020; 12(7):693. https://doi.org/10.3390/v12070693
Chicago/Turabian StyleBarrett, Chelsea T., and Rebecca Ellis Dutch. 2020. "Viral Membrane Fusion and the Transmembrane Domain" Viruses 12, no. 7: 693. https://doi.org/10.3390/v12070693
APA StyleBarrett, C. T., & Dutch, R. E. (2020). Viral Membrane Fusion and the Transmembrane Domain. Viruses, 12(7), 693. https://doi.org/10.3390/v12070693