SETDB1-Mediated Silencing of Retroelements

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Regulation of Retroelement Expression by H3K9 and DNA Methylation
3. Characterization of SETDB1
4. SETDB1 Regulates Retroelements Also in Somatic Cells
5. In Vivo Function of SETDB1
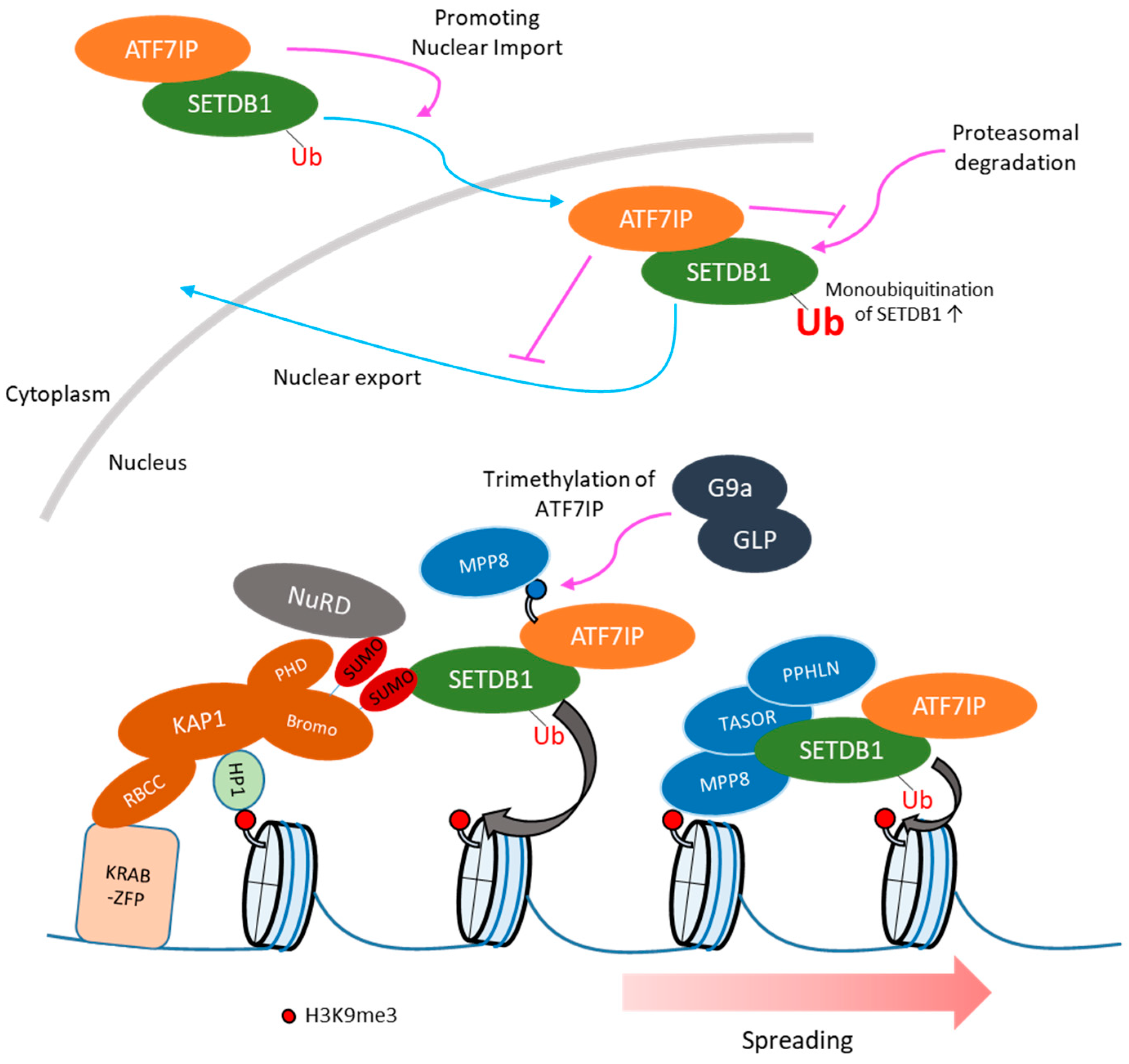
6. Regulation of Subcellular Localization of SETDB1
7. SETDB1-Mediated Retroelement Silencing: Recruitment of SETDB1 to Retroelements
8. SETDB1-Mediated Retroelement Silencing: Spreading of H3K9me3 from KAP1 Binding Sites
9. SETDB1-Mediated Retroelement Silencing: Maintenance of Heterochromatin during Cell Division
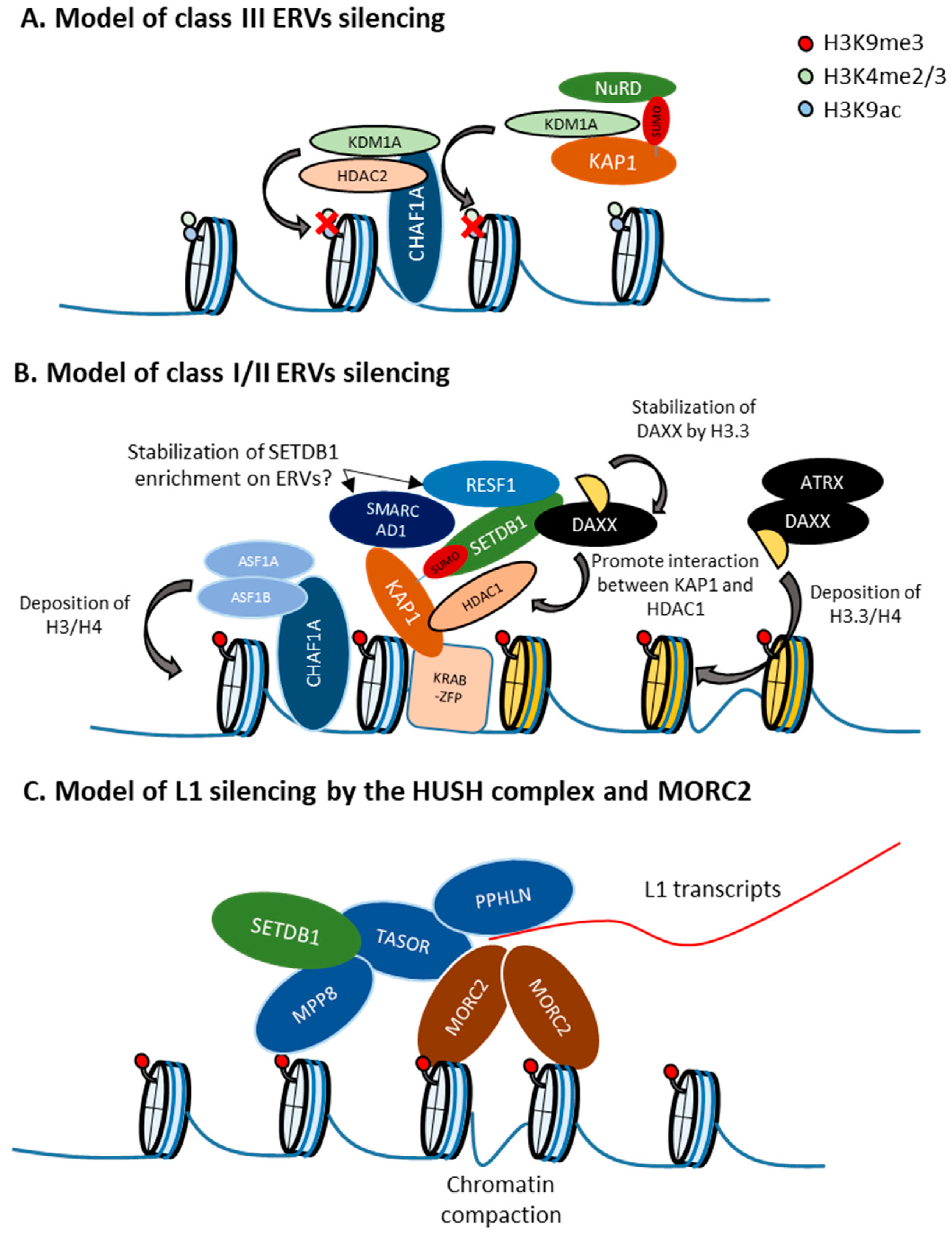
10. Retroelement-Type Specific Regulation
11. Concluding Remarks
Funding
Conflicts of Interest
References
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequence and comparative analysis of the cat genome. Nature 2002, 420, 520–562. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar]
- Maksakova, I.A.; Romanish, M.T.; Gagnier, L.; Dunn, C.A.; Van De Lagemaat, L.N.; Mager, D.L. Retroviral Elements and Their Hosts: Insertional Mutagenesis in the Mouse Germ Line. PLoS Genet. 2006, 2, e2. [Google Scholar] [CrossRef]
- Huang, C.R.L.; Burns, K.H.; Boeke, J.D. Active Transposition in Genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr. Mobile Elements: Drivers of Genome Evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Burns, K.H. Mobilizing Diversity: Transposable Element Insertions in Genetic Variation and Disease. Mob. DNA 2010, 1, 21. [Google Scholar] [CrossRef]
- Mager, D.L.; Stoye, J.P. Mammalian Endogenous Retroviruses. Microbiol. Spectr. 2015, 3, 1079–1100. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting Retrotransposons: A Review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef]
- Groh, S.; Schotta, G. Silencing of Endogenous Retroviruses by Heterochromatin. Cell Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral Silencing in Embryonic Stem Cells Requires the Histone Methyltransferase Eset. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Goyal, P.; Bullwinkel, J.; Brown, J.P.; Bilenky, M.; Mager, D.L.; Singh, P.B.; Lorincz, M.C. H3k9me3-Binding Proteins Are Dispensable for Setdb1/H3k9me3-Dependent Retroviral Silencing. Epigenetics Chromatin 2011, 4, 12. [Google Scholar] [CrossRef]
- Zhang, Z.; Harrison, P.M.; Liu, Y.; Gerstein, M. Millions of Years of Evolution Preserved: A Comprehensive Catalog of the Processed Pseudogenes in the Human Genome. Genome Res. 2003, 13, 2541–2558. [Google Scholar] [CrossRef]
- Ferguson-Smith, A.C.; Sasaki, H.; Cattanach, B.M.; Surani, M.A. Parental-Origin-Specific Epigenetic Modification of the Mouse H19 Gene. Nature 1993, 362, 751–755. [Google Scholar] [CrossRef]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA Methylation in Genomic Imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Webber, A.L.; Tilghman, S.M.; Brunkow, M.E. Epigenetic Mechanisms Underlying the Imprinting of the Mouse H19 Gene. Genes Dev. 1993, 7, 1663–1673. [Google Scholar] [CrossRef]
- Stöger, R. Maternal-Specific Methylation of the Imprinted Mouse Igf2r Locus Identifies the Expressed Locus as Carrying the Imprinting Signal. Cell 1993, 73, 61–71. [Google Scholar] [CrossRef]
- Mohandas, T.; Sparkes, R.; Shapiro, L. Reactivation of an Inactive Human X Chromosome: Evidence for X Inactivation by DNA Methylation. Science 1981, 211, 393–396. [Google Scholar] [CrossRef]
- Lock, L.F.; Takagi, N.; Martin, G.R. Methylation of the Hprt Gene on the Inactive X Occurs after Chromosome Inactivation. Cell 1987, 48, 39–46. [Google Scholar] [CrossRef]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of Iap Endogenous Retroviruses Is Constrained by Cytosine Methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef]
- Hutnick, L.K.; Huang, X.; Loo, T.-C.; Ma, Z.; Fan, G. Repression of Retrotransposal Elements in Mouse Embryonic Stem Cells Is Primarily Mediated by a DNA Methylation-independent Mechanism. J. Boil. Chem. 2010, 285, 21082–21091. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.-W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of Chromatin Structure by Site-Specific Histone H3 Methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Nakayama, J.-I.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef]
- Martens, J.H.A.; O’Sullivan, R.J.; Braunschweig, U.; Opravil, S.; Radolf, M.; Steinlein, P.; Jenuwein, T. The Profile of Repeat-Associated Histone Lysine Methylation States in the Mouse Epigenome. EMBO J. 2005, 24, 800–812. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. Setdb1: A Novel Kap-1-Associated Histone H3, Lysine 9-Specific Methyltransferase That Contributes to Hp1-Mediated Silencing of Euchromatic Genes by Krab Zinc-Finger Proteins. Genome Res. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. SET Domain-containing Protein, G9a, Is a Novel Lysine-preferring Mammalian Histone Methyltransferase with Hyperactivity and Specific Selectivity to Lysines 9 and 27 of Histone H3. J. Boil. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a Histone Methyltransferase Plays a Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and Is Essential for Early Embryogenesis. Genome Res. 2002, 16, 1779–1791. [Google Scholar] [CrossRef]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone Methyltransferases G9a and Glp Form Heteromeric Complexes and Are Both Crucial for Methylation of Euchromatin at H3-K9. Genome Res. 2005, 19, 815–826. [Google Scholar] [CrossRef]
- Karimi, M.; Goyal, P.M.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA Methylation and Setdb1/H3k9me3 Regulate Predominantly Distinct Sets of Genes, Retroelements, and Chimeric Transcripts in Mescs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Thompson, P.; Goyal, P.; Jones, S.J.M.; Singh, P.B.; Karimi, M.M.; Lorincz, M.C. Distinct Roles of Kap1, Hp1 and G9a/Glp in Silencing of the Two-Cell-Specific Retrotransposon Mervl in Mouse Es Cells. Epigenetics Chromatin 2013, 6, 15. [Google Scholar] [CrossRef]
- Bulut-Karslioglu, A.; De La Rosa-Velázquez, I.A.; Ramírez, F.; Barenboim, M.; Onishi-Seebacher, M.; Arand, J.; Galán, C.; Winter, G.E.; Engist, B.; Gerle, B.; et al. Suv39h-Dependent H3K9me3 Marks Intact Retrotransposons and Silences LINE Elements in Mouse Embryonic Stem Cells. Mol. Cell 2014, 55, 277–290. [Google Scholar] [CrossRef]
- Yang, L.; Xia, L.; Wu, D.Y.; Wang, H.; Chansky, H.A.; Schubach, W.H.; Hickstein, D.D.; Zhang, Y. Molecular Cloning of Eset, a Novel Histone H3-Specific Methyltransferase That Interacts with Erg Transcription Factor. Oncogene 2002, 21, 148–152. [Google Scholar] [CrossRef]
- Poulin, G.; Dong, Y.; Fraser, A.G.; Hopper, N.A.; Ahringer, J. Chromatin Regulation and Sumoylation in the Inhibition of Ras-Induced Vulval Development in Caenorhabditis Elegans. EMBO J. 2005, 24, 2613–2623. [Google Scholar] [CrossRef]
- Stabell, M.; Bjørkmo, M.; Aalen, R.B.; Lambertsson, A. The Drosophila Set Domain Encoding Gene Deset Is Essential for Proper Development. Hereditas 2006, 143, 177–188. [Google Scholar] [CrossRef]
- Chen, C.; Nott, T.; Jin, J.; Pawson, T. Deciphering Arginine Methylation: Tudor Tells the Tale. Nat. Rev. Mol. Cell Boil. 2011, 12, 629–642. [Google Scholar] [CrossRef]
- Lu, R.; Wang, G.G. Tudor: A Versatile Family of Histone Methylation ‘Readers’. Trends Biochem. Science 2013, 38, 546–555. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Qin, S.; Kungulovski, G.; Tempel, W.; Liu, Y.; Bashtrykov, P.; Stiefelmaier, J.; Jurkowski, T.; Kudithipudi, S.; Weirich, S.; et al. H3k14ac Is Linked to Methylation of H3k9 by the Triple Tudor Domain of Setdb1. Nat. Commun. 2017, 8, 2057. [Google Scholar] [CrossRef]
- Price, A.J.; Manjegowda, M.C.; Kain, J.; Anandh, S.; Bochkis, I.M. Hdac3, Setdb1, and Kap1 Mark H3k9me3/H3k14ac Bivalent Regions in Young and Aged Liver. Aging Cell 2020, 19, e13092. [Google Scholar] [CrossRef]
- Matsumura, Y.; Nakaki, R.; Inagaki, T.; Yoshida, A.; Kano, Y.; Kimura, H.; Tanaka, T.; Tsutsumi, S.; Nakao, M.; Doi, T.; et al. H3k4/H3k9me3 Bivalent Chromatin Domains Targeted by Lineage-Specific DNA Methylation Pauses Adipocyte Differentiation. Mol. Cell 2015, 60, 584–596. [Google Scholar] [CrossRef]
- Sun, L.; Fang, J. E3-Independent Constitutive Monoubiquitination Complements Histone Methyltransferase Activity of Setdb1. Mol. Cell 2016, 62, 958–966. [Google Scholar] [CrossRef]
- Ishimoto, K.; Kawamata, N.; Uchihara, Y.; Okubo, M.; Fujimoto, R.; Gotoh, E.; Kakinouchi, K.; Mizohata, E.; Hino, N.; Okada, Y.; et al. Ubiquitination of Lysine 867 of the Human Setdb1 Protein Upregulates Its Histone H3 Lysine 9 (H3k9) Methyltransferase Activity. PLoS ONE 2016, 11, e0165766. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. Ph.D. Domain-Mediated E3 Ligase Activity Directs Intramolecular Sumoylation of an Adjacent Bromodomain Required for Gene Silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Park, J.S.; Kang, Y.-K. Regulated Nuclear Entry of over-Expressed Setdb1. Genes Cells 2013, 18, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.L.; Chansky, H.A.; Zielinska-Kwiatkowska, A.; Matsui, Y.; Yang, L. Genomic Structure and Expression of the Mouse Eset Gene Encoding an Erg-Associated Histone Methyltransferase with a Set Domain. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2003, 1629, 8–14. [Google Scholar] [CrossRef]
- Dodge, J.E.; Kang, Y.-K.; Beppu, H.; Lei, H.; Li, E. Histone H3-K9 Methyltransferase ESET Is Essential for Early Development. Mol. Cell. Boil. 2004, 24, 2478–2486. [Google Scholar] [CrossRef]
- Liu, S.; Brind’Amour, J.; Karimi, M.M.; Shirane, K.; Bogutz, A.; Lefebvre, L.; Sasaki, H.; Shinkai, Y.; Lorincz, M.C. Setdb1 Is Required for Germline Development and Silencing of H3k9me3-Marked Endogenous Retroviruses in Primordial Germ Cells. Genes Dev. 2014, 28, 2041–2055. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic Reprogramming in Mammalian Development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Robbez-Masson, L.; Tie, C.H.; Conde, L.; Tunbak, H.; Husovsky, C.; Tchasovnikarova, I.A.; Timms, R.T.; Herrero, J.; Lehner, P.J.; Rowe, H.M. The Hush Complex Cooperates with Trim28 to Repress Young Retrotransposons and New Genes. Genome Res. 2018, 28, 836–845. [Google Scholar] [CrossRef]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. Kap1 Controls Endogenous Retroviruses in Embryonic Stem Cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Collins, P.L.; Kyle, K.E.; Egawa, T.; Shinkai, Y.; Oltz, E.M. The Histone Methyltransferase Setdb1 Represses Endogenous and Exogenous Retroviruses in B Lymphocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 8367–8372. [Google Scholar] [CrossRef]
- Takikita, S.; Muro, R.; Takai, T.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Oda, H.; Kitajima, M.; Oshima, K.; Hattori, M.; et al. A Histone Methyltransferase ESET Is Critical for T Cell Development. J. Immunol. 2016, 197, 2269–2279. [Google Scholar] [CrossRef]
- Tan, S.-L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential Roles of the Histone Methyltransferase Eset in the Epigenetic Control of Neural Progenitor Cells During Development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Takemoto, K.; Shinkai, Y. A Somatic Role for the Histone Methyltransferase Setdb1 in Endogenous Retrovirus Silencing. Nat. Commun. 2018, 9, 1683. [Google Scholar] [CrossRef]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.P.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. Kap1 Regulates Endogenous Retroviruses in Adult Human Cells and Contributes to Innate Immune Control. EMBO Rep. 2018, 19, e45000. [Google Scholar] [CrossRef] [PubMed]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their Krab-Zfp Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Yang, P.; Füchtbauer, A.C.; Füchtbauer, E.-M.; Silva, A.M.; Park, C.; Wu, W.; Nielsen, A.L.; Pedersen, F.S.; Macfarlan, T.S. The Krab Zinc Finger Protein Zfp809 Is Required to Initiate Epigenetic Silencing of Endogenous Retroviruses. Genome Res. 2015, 29, 538–554. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Zhang, X.; Qin, J.; Wan, Y.; Hu, Y.; Liu, T.; Li, J.; Dong, W.; Du, E.; Pan, C.; et al. The histone methyltransferase ESET is required for the survival of spermatogonial stem/progenitor cells in mice. Cell Death Dis. 2014, 5, e1196. [Google Scholar] [CrossRef]
- Eymery, A.; Liu, Z.; Ozonov, E.A.; Stadler, M.B.; Peters, A.H.F.M. The Methyltransferase Setdb1 Is Essential for Meiosis and Mitosis in Mouse Oocytes and Early Embryos. Development 2016, 143, 2767–2779. [Google Scholar] [CrossRef]
- Lawson, K.A.; Teteak, C.J.; Gao, J.; Li, N.; Hacquebord, J.; Ghatan, A.; Zielinska-Kwiatkowska, A.; Song, G.; Chansky, H.A.; Yang, L. Eset Histone Methyltransferase Regulates Osteoblastic Differentiation of Mesenchymal Stem Cells During Postnatal Bone Development. FEBS Lett. 2013, 587, 3961–3967. [Google Scholar] [CrossRef]
- Yang, L.; Lawson, K.; Teteak, C.J.; Zou, J.; Hacquebord, J.; Patterson, D.; Ghatan, A.C.; Mei, Q.; Zielinska-Kwiatkowska, A.; Bain, S.D.; et al. Eset Histone Methyltransferase Is Essential to Hypertrophic Differentiation of Growth Plate Chondrocytes and Formation of Epiphyseal Plates. Dev. Boil. 2013, 380, 99–110. [Google Scholar] [CrossRef]
- Lawson, K.; Teteak, C.J.; Zou, J.; Hacquebord, J.; Ghatan, A.; Zielinska-Kwiatkowska, A.; Fernandes, R.J.; Chansky, H.A.; Yang, L. Mesenchyme-Specific Knockout of Eset Histone Methyltransferase Causes Ectopic Hypertrophy and Terminal Differentiation of Articular Chondrocytes. J. Boil. Chem. 2013, 288, 32119–32125. [Google Scholar] [CrossRef]
- Pasquarella, A.; Ebert, A.; De Almeida, G.P.; Hinterberger, M.; Kazerani, M.; Nuber, A.; Ellwart, J.; Klein, L.; Busslinger, M.; Schotta, G. Retrotransposon Derepression Leads to Activation of the Unfolded Protein Response and Apoptosis in Pro-B Cells. Development 2016, 143, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Koide, S.; Oshima, M.; Takubo, K.; Yamazaki, S.; Nitta, E.; Saraya, A.; Aoyama, K.; Kato, Y.; Miyagi, S.; Nakajima-Takagi, Y.; et al. Setdb1 Maintains Hematopoietic Stem and Progenitor Cells by Restricting the Ectopic Activation of Nonhematopoietic Genes. Blood 2016, 128, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Herzner, A.-M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of Retrotransposons by Setdb1 Inhibits the Interferon Response in Acute Myeloid Leukemia. J. Cell Boil. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.-Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.-H.; et al. Gut Stem Cell Necroptosis by Genome Instability Triggers Bowel Inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Gotoh, E.; Kawamata, N.; Ishimoto, K.; Uchihara, Y.; Iwanari, H.; Sugiyama, A.; Kawamura, T.; Mochizuki, Y.; Tanaka, T.; et al. Analysis of the Subcellular Localization of the Human Histone Methyltransferase Setdb1. Biochem. Biophys. Res. Commun. 2015, 465, 725–731. [Google Scholar] [CrossRef]
- Wang, H.; An, W.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Chatton, B.; Tempst, P.; Roeder, R.G.; Zhang, Y. Mam Facilitates Conversion by Eset of Dimethyl to Trimethyl Lysine 9 of Histone H3 to Cause Transcriptional Repression. Mol. Cell 2003, 12, 475–487. [Google Scholar] [CrossRef]
- Ichimura, T.; Watanabe, S.; Sakamoto, Y.; Aoto, T.; Fujita, N.; Nakao, M. Transcriptional Repression and Heterochromatin Formation by Mbd1 and Mcaf/Am Family Proteins. J. Boil. Chem. 2005, 280, 13928–13935. [Google Scholar] [CrossRef]
- Minkovsky, A.; Sahakyan, A.; Rankin-Gee, E.; Bonora, G.; Patel, S.G.; Plath, K. The Mbd1-Atf7ip-Setdb1 Pathway Contributes to the Maintenance of X Chromosome Inactivation. Epigenetics Chromatin 2014, 7, 12. [Google Scholar] [CrossRef]
- Timms, R.T.; Tchasovnikarova, I.A.; Antrobus, R.; Dougan, G.; Lehner, P.J. Atf7ip-Mediated Stabilization of the Histone Methyltransferase Setdb1 Is Essential for Heterochromatin Formation by the Hush Complex. Cell Rep. 2016, 17, 653–659. [Google Scholar] [CrossRef]
- Tsusaka, T.; Shimura, C.; Shinkai, Y. Atf7ip Regulates Setdb1 Nuclear Localization and Increases Its Ubiquitination. EMBO Rep. 2019, 20, 48297. [Google Scholar] [CrossRef]
- Fukuda, K.; Okuda, A.; Yusa, K.; Shinkai, Y. A Crispr Knockout Screen Identifies Setdb1-Target Retroelement Silencing Factors in Embryonic Stem Cells. Genome Res. 2018, 28, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.M.; Honemann-Capito, M.; Egger-Adam, D.; Wodarz, A. Windei, the Drosophila Homolog of Mam/Mcaf1, Is an Essential Cofactor of the H3k9 Methyl Transferase Dsetdb1/Eggless in Germ Line Development. PLoS Genet. 2009, 5, e1000644. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, B.; Chen, H.-M.; Moresco, J.J.; Orelo, B.D.; Yang, B.; Gaspar, J.M.; Keppler-Ross, S.; Yates, I.J.R.; Hall, D.H.; Maine, E.M.; et al. Regulated nuclear accumulation of a histone methyltransferase times the onset of heterochromatin formation inC. elegansembryos. Sci. Adv. 2018, 4, eaat6224. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.E.; Methot, S.P.; Guidi, M.; Katic, I.; Gasser, S.M.; Padeken, J. Heterochromatic Foci and Transcriptional Repression by an Unstructured Met-2/Setdb1 Co-Factor Lin-65. J. Cell Biol. 2019, 218, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Osumi, K.; Sato, K.; Murano, K.; Siomi, H.; Siomi, M.C. Essential Roles of Windei and Nuclear Monoubiquitination of Eggless/Setdb1 in Transposon Silencing. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Matheson, N.J.; Wals, K.; Antrobus, R.; Göttgens, B.; Dougan, G.; Dawson, M.A.; Lehner, P.J. Gene Silencing. Epigenetic Silencing by the Hush Complex Mediates Position-Effect Variegation in Human Cells. Science 2015, 348, 1481–1485. [Google Scholar] [CrossRef]
- Tsusaka, T.; Kikuchi, M.; Shimazu, T.; Suzuki, T.; Sohtome, Y.; Akakabe, M.; Sodeoka, M.; Dohmae, N.; Umehara, T.; Shinkai, Y. Tri-Methylation of Atf7ip by G9a/Glp Recruits the Chromodomain Protein Mpp8. Epigenetics Chromatin 2018, 11, 56. [Google Scholar] [CrossRef]
- Friedman, J.; Fredericks, W.J.; Speicher, D.W.; Huang, X.P.; Jensen, D.E.; Neilson, E.G.; Rauscher, F.J. Kap-1, a Novel Corepressor for the Highly Conserved Krab Repression Domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef]
- Peng, H.; Begg, G.E.; Schultz, D.C.; Friedman, J.; Jensen, D.E.; Speicher, D.W.; Rauscher, F.J. Reconstitution of the Krab-Kap-1 Repressor Complex: A Model System for Defining the Molecular Anatomy of Ring-B Box-Coiled-Coil Domain-Mediated Protein-Protein Interactions. J. Mol. Boil. 2000, 295, 1139–1162. [Google Scholar] [CrossRef]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J. The Phd and Bromodomains of Kap-1 Form a Cooperative Unit That Recruits a Novel Isoform of the Mi-2alpha Subunit of Nurd. Genome Res. 2001, 15, 428–443. [Google Scholar] [CrossRef]
- Zeng, L.; Yap, K.L.; Ivanov, A.V.; Wang, X.; Mujtaba, S.; Plotnikova, O.; Rauscher, F.J.; Zhou, M.-M. Structural Insights into Human Kap1 Phd Finger-Bromodomain and Its Role in Gene Silencing. Nat. Struct. Mol. Boil. 2008, 15, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Farnham, P.J. Kap1 Protein: An Enigmatic Master Regulator of the Genome. J. Boil. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef] [PubMed]
- Huntley, S.; Baggott, D.M.; Hamilton, A.T.; Tran-Gyamfi, M.; Yang, S.; Kim, J.; Gordon, L.; Branscomb, E.; Stubbs, L. A Comprehensive Catalog of Human Krab-Associated Zinc Finger Genes: Insights into the Evolutionary History of a Large Family of Transcriptional Repressors. Genome Res. 2006, 16, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Birtle, Z.; Ponting, C.P. Meisetz and the Birth of the Krab Motif. Bioinformatics 2006, 22, 2841–2845. [Google Scholar] [CrossRef]
- Thomas, J.H.; Schneider, S. Coevolution of Retroelements and Tandem Zinc Finger Genes. Genome Res. 2011, 21, 1800–1812. [Google Scholar] [CrossRef]
- Jacobs, F.M.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An Evolutionary Arms Race between Krab Zinc-Finger Genes Znf91/93 and Sva/L1 Retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef]
- Imbeault, M.; Helleboid, P.-Y.; Trono, D. Coevolution of Retroelements and Tandem Zinc Finger Genes. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef]
- Wolf, D.; Goff, S.P. Embryonic Stem Cells Use Zfp809 to Silence Retroviral Dnas. Nature 2009, 458, 1201–1204. [Google Scholar] [CrossRef]
- Castro-Díaz, N.; Ecco, G.; Coluccio, A.; Kapopoulou, A.; Yazdanpanah, B.; Friedli, M.; Duc, J.; Jang, S.M.; Turelli, P.; Trono, D. Evolutionally Dynamic L1 Regulation in Embryonic Stem Cells. Genes Dev. 2014, 28, 1397–1409. [Google Scholar] [CrossRef]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. Krab-Zinc Finger Proteins and Kap1 Can Mediate Long-Range Transcriptional Repression through Heterochromatin Spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Thompson, P.; Dulberg, V.; Moon, K.-M.; Foster, L.J.; Chen, C.; Karimi, M.M.; Lorincz, M.C. hnRNP K Hnrnp K Coordinates Transcriptional Silencing by Setdb1 in Embryonic Stem Cells. PLoS Genet. 2015, 11, e1004933. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.M.; Kapopoulou, A.; Corsinotti, A.; Fasching, L.; Macfarlan, T.S.; Tarabay, Y.; Viville, S.; Jakobsson, J.; Pfaff, S.L.; Trono, D. Trim28 Repression of Retrotransposon-Based Enhancers Is Necessary to Preserve Transcriptional Dynamics in Embryonic Stem Cells. Genome Res. 2012, 23, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Ball, A.R., Jr.; Yokomori, K.; Ball, A.R. Hp1: Heterochromatin Binding Proteins Working the Genome. Epigenetics 2010, 5, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Zegerman, P.; Partridge, J.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective Recognition of Methylated Lysine 9 on Histone H3 by the Hp1 Chromo Domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of Histone H3 Lysine 9 Creates a Binding Site for Hp1 Proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Kourmouli, N.; Sun, Y.-M.; van der Sar, S.; Singh, P.B.; Brown, J.P. Epigenetic Regulation of Mammalian Pericentric Heterochromatin in Vivo by Hp1. Biochem. Biophys. Res. Commun. 2005, 337, 901–907. [Google Scholar] [CrossRef]
- Ostapcuk, V.; Mohn, F.; Carl, S.; Basters, A.; Hess, D.; Iesmantavicius, V.; Lampersberger, L.; Flemr, M.; Pandey, A.; Thomä, N.H.; et al. Activity-Dependent Neuroprotective Protein Recruits Hp1 and Chd4 to Control Lineage-Specifying Genes. Nature 2018, 557, 739–743. [Google Scholar] [CrossRef]
- Kokura, K.; Sun, L.; Bedford, M.T.; Fang, J. Methyl-H3k9-Binding Protein Mpp8 Mediates E-Cadherin Gene Silencing and Promotes Tumour Cell Motility and Invasion. EMBO J. 2010, 29, 3673–3687. [Google Scholar] [CrossRef]
- Bua, D.; Kuo, A.J.; Cheung, P.; Liu, C.L.; Migliori, V.; Espejo, A.; Casadio, F.; Bassi, C.; Amati, B.; Bedford, M.T.; et al. Epigenome Microarray Platform for Proteome-Wide Dissection of Chromatin-Signaling Networks. PLoS ONE 2009, 4, e6789. [Google Scholar] [CrossRef]
- Liu, H.; Galka, M.; Iberg, A.; Wang, Z.; Li, L.; Voss, C.; Jiang, X.; Lajoie, G.; Huang, Z.; Bedford, M.T.; et al. Systematic Identification of Methyllysine-Driven Interactions for Histone and Nonhistone Targets. J. Proteome Res. 2010, 9, 5827–5836. [Google Scholar] [CrossRef]
- Kim, J.; Daniel, J.; Espejo, A.; Lake, A.; Krishna, M.; Xia, L.; Zhang, Y.; Bedford, M.T. Tudor, Mbt and Chromo Domains Gauge the Degree of Lysine Methylation. EMBO Rep. 2006, 7, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, P.; Westbrook, T.F.; Ottinger, M.; Pavlova, N.; Chang, B.; Macia, E.; Shi, Y.-J.; Barretina, J.; Liu, J.; Howley, P.; et al. Cdyl Bridges Rest and Histone Methyltransferases for Gene Repression and Suppression of Cellular Transformation. Mol. Cell 2008, 32, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Franz, H.; Mosch, K.; Soeroes, S.; Urlaub, H.; Fischle, W. Multimerization and H3k9me3 Binding Are Required for Cdyl1b Heterochromatin Association. J. Boil. Chem. 2009, 284, 35049–35059. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective Silencing of Euchromatic L1s Revealed by Genome-Wide Screens for L1 Regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef]
- Timms, R.T.; Tchasovnikarova, I.A.; Lehner, P.J. Position-Effect Variegation Revisited: Hushing up Heterochromatin in Human Cells. BioEssays 2016, 38, 333–343. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3k9 Methylation Is a Barrier During Somatic Cell Reprogramming into Ipscs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef]
- Cheloufi, S.; Elling, U.; Hopfgartner, B.; Jung, Y.L.; Murn, J.; Ninova, M.; Hubmann, M.; Badeaux, A.I.; Ang, C.E.; Tenen, D.; et al. The Histone Chaperone Caf-1 Safeguards Somatic Cell Identity. Nature 2015, 528, 218–224. [Google Scholar] [CrossRef]
- Smith, S.; Stillman, B. Stepwise Assembly of Chromatin During DNA Replication In Vitro. EMBO J. 1991, 10, 971–980. [Google Scholar] [CrossRef]
- Quivy, J.-P.; Roche, D.; Kirschner, D.; Tagami, H.; Nakatani, Y.; Almouzni, G. A Caf-1 Dependent Pool of Hp1 During Heterochromatin Duplication. EMBO J. 2004, 23, 3516–3526. [Google Scholar] [CrossRef]
- Houlard, M.; Berlivet, S.; Probst, A.; Quivy, J.-P.; Héry, P.; Almouzni, G.; Gerard, M. Caf-1 Is Essential for Heterochromatin Organization in Pluripotent Embryonic Cells. PLoS Genet. 2006, 2, e181. [Google Scholar] [CrossRef]
- Yang, B.X.; El Farran, C.; Guo, H.C.; Yu, T.; Fang, H.T.; Wang, H.F.; Schlesinger, S.; Seah, S.; Goh, G.Y.L.; Neo, S.P.; et al. Systematic Identification of Factors for Provirus Silencing in Embryonic Stem Cells. Cell 2015, 163, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Schoorlemmer, J.; Perez-Palacios, R.; Climent, M.; Guallar, D.; Muniesa, P. Regulation of Mouse Retroelement Muerv-L/Mervl Expression by Rex1 and Epigenetic Control of Stem Cell Potency. Front. Oncol. 2014, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Sachs, P.; Ding, D.; Bergmaier, P.; Lamp, B.; Schlagheck, C.; Finkernagel, F.; Nist, A.; Stiewe, T.; Mermoud, J.E. Smarcad1 Atpase Activity Is Required to Silence Endogenous Retroviruses in Embryonic Stem Cells. Nat. Commun. 2019, 10, 1335. [Google Scholar] [CrossRef] [PubMed]
- Elsässer, S.; Noh, K.-M.; Diaz, N.; Allis, C.D.; Banaszynski, L. Histone H3.3 Is Required for Endogenous Retroviral Element Silencing in Embryonic Stem Cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Hoelper, D.; Huang, H.; Jain, A.; Patel, D.J.; Lewis, P.W. Structural and Mechanistic Insights into Atrx-Dependent and -Independent Functions of the Histone Chaperone Daxx. Nat. Commun. 2017, 8, 1193. [Google Scholar] [CrossRef] [PubMed]
- Prigozhin, D.M.; Albecka, A.; Douse, C.H.; Tchasovnikarova, I.A.; Timms, R.T.; Farleigh, L.E.; Oda, S.; Freung, S.M.V.; Maslen, S.; Lehner, P.J.; et al. Periphilin self-association underpins epigenetic silencing by the HUSH complex. bioRxiv 2019. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Douse, C.H.; Roberts, R.; Dougan, G.; Kingston, R.E.; Modis, Y.; Lehner, P.J. Hyperactivation of Hush Complex Function by Charcot-Marie-Tooth Disease Mutation in Morc2. Nat. Genet. 2017, 49, 1035–1044. [Google Scholar] [CrossRef]
- Douse, C.H.; Bloor, S.; Liu, Y.; Shamin, M.; Tchasovnikarova, I.A.; Timms, R.T.; Lehner, P.J.; Modis, Y. Neuropathic Morc2 Mutations Perturb Ghkl Atpase Dimerization Dynamics and Epigenetic Silencing by Multiple Structural Mechanisms. Nat. Commun. 2018, 9, 651. [Google Scholar] [CrossRef]
- Wolf, G.; Rebollo, R.; Karimi, M.M.; Ewing, A.; Kamada, R.; Wu, W.; Wu, B.; Bachu, M.; Ozato, K.; Faulkner, G.J.; et al. On the Role of H3.3 in Retroviral Silencing. Nature 2017, 548, E1–E3. [Google Scholar] [CrossRef]
- Elsasser, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Elsasser Et Al. Reply. Nature 2017, 548, E7–E9. [Google Scholar] [CrossRef]
- Sadic, D.; Schmidt, K.; Groh, S.; Kondofersky, I.; Ellwart, J.; Fuchs, C.; Theis, F.J.; Schotta, G. Atrx Promotes Heterochromatin Formation at Retrotransposons. EMBO Rep. 2015, 16, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Tamas, R.; Bock, I.; Tattermusch, A.; Dimitrova, E.; Kudithipudi, S.; Ragozin, S.; Jeltsch, A. The Atrx-Add Domain Binds to H3 Tail Peptides and Reads the Combined Methylation State of K4 and K9. Hum. Mol. Genet. 2011, 20, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Song, Y.; Chen, W.; Wang, X.; Miao, Z.; Cao, L.; Li, F.; Wang, G. By Recruiting Hdac1, Morc2 Suppresses P21 Waf1/Cip1 in Gastric Cancer. Oncotarget 2015, 6, 16461–16470. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Li, Y.; Zhang, J.; Liu, D.; Liu, F.; Zhao, Y.; Shen, T.; Li, F. Involvement of Histone Deacetylation in Morc2-Mediated Down-Regulation of Carbonic Anhydrase Ix. Nucleic Acids Res. 2010, 38, 2813–2824. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuda, K.; Shinkai, Y. SETDB1-Mediated Silencing of Retroelements. Viruses 2020, 12, 596. https://doi.org/10.3390/v12060596
Fukuda K, Shinkai Y. SETDB1-Mediated Silencing of Retroelements. Viruses. 2020; 12(6):596. https://doi.org/10.3390/v12060596
Chicago/Turabian StyleFukuda, Kei, and Yoichi Shinkai. 2020. "SETDB1-Mediated Silencing of Retroelements" Viruses 12, no. 6: 596. https://doi.org/10.3390/v12060596
APA StyleFukuda, K., & Shinkai, Y. (2020). SETDB1-Mediated Silencing of Retroelements. Viruses, 12(6), 596. https://doi.org/10.3390/v12060596