Unexpected Genetic Diversity of Two Novel Swine MRVs in Italy

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. MRVs Detection in One Single Swine Farm in North East Italy
2.2. Virus Isolation in Cell Cultures
2.3. Reconstruction of MRV Genomes
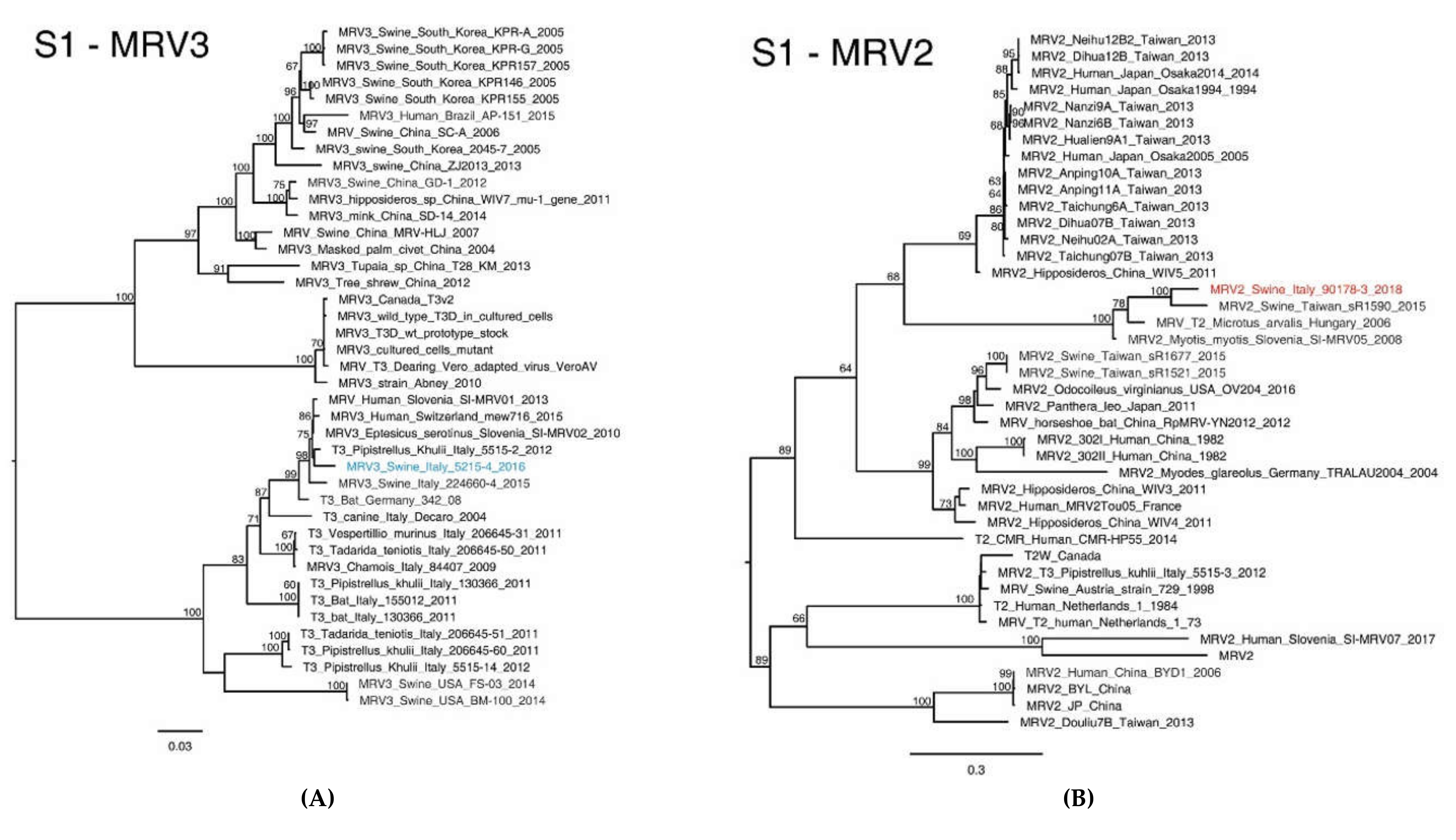
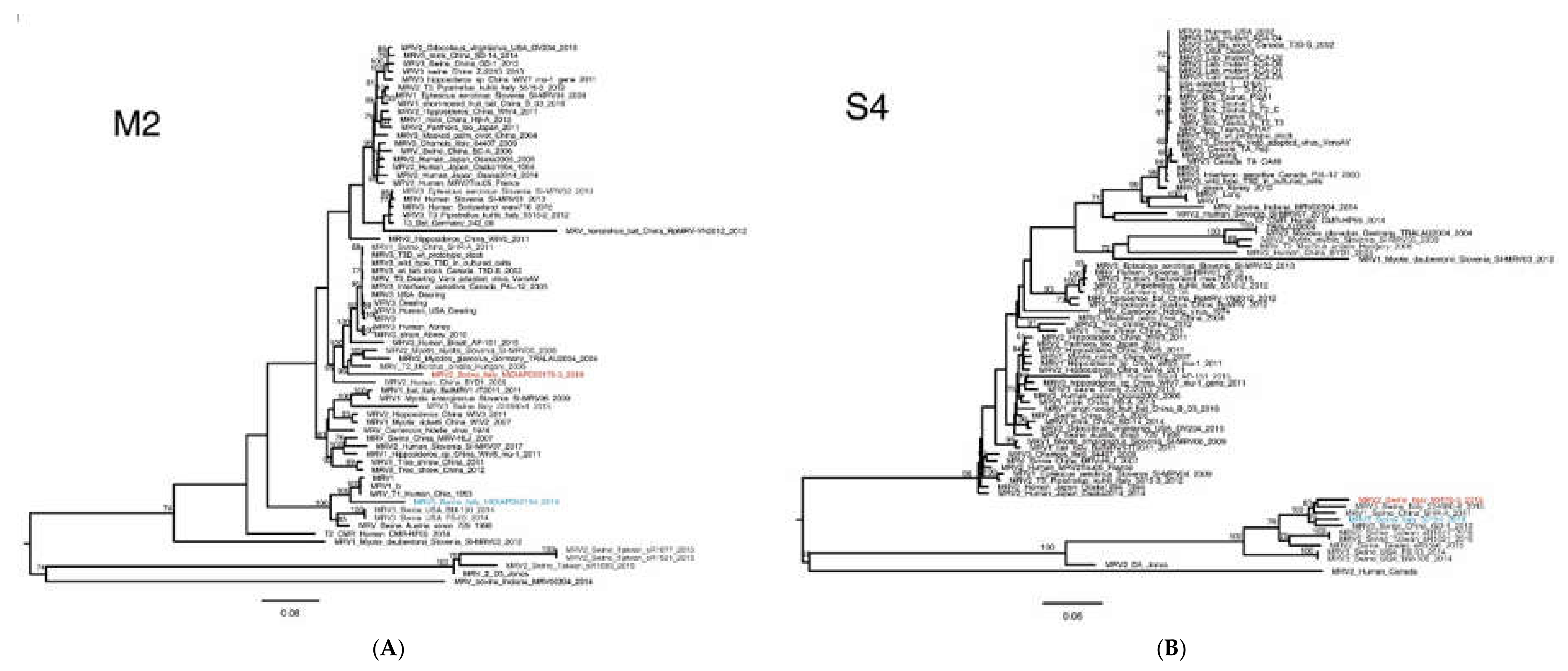
2.4. MRV Phylogenetic Analysis
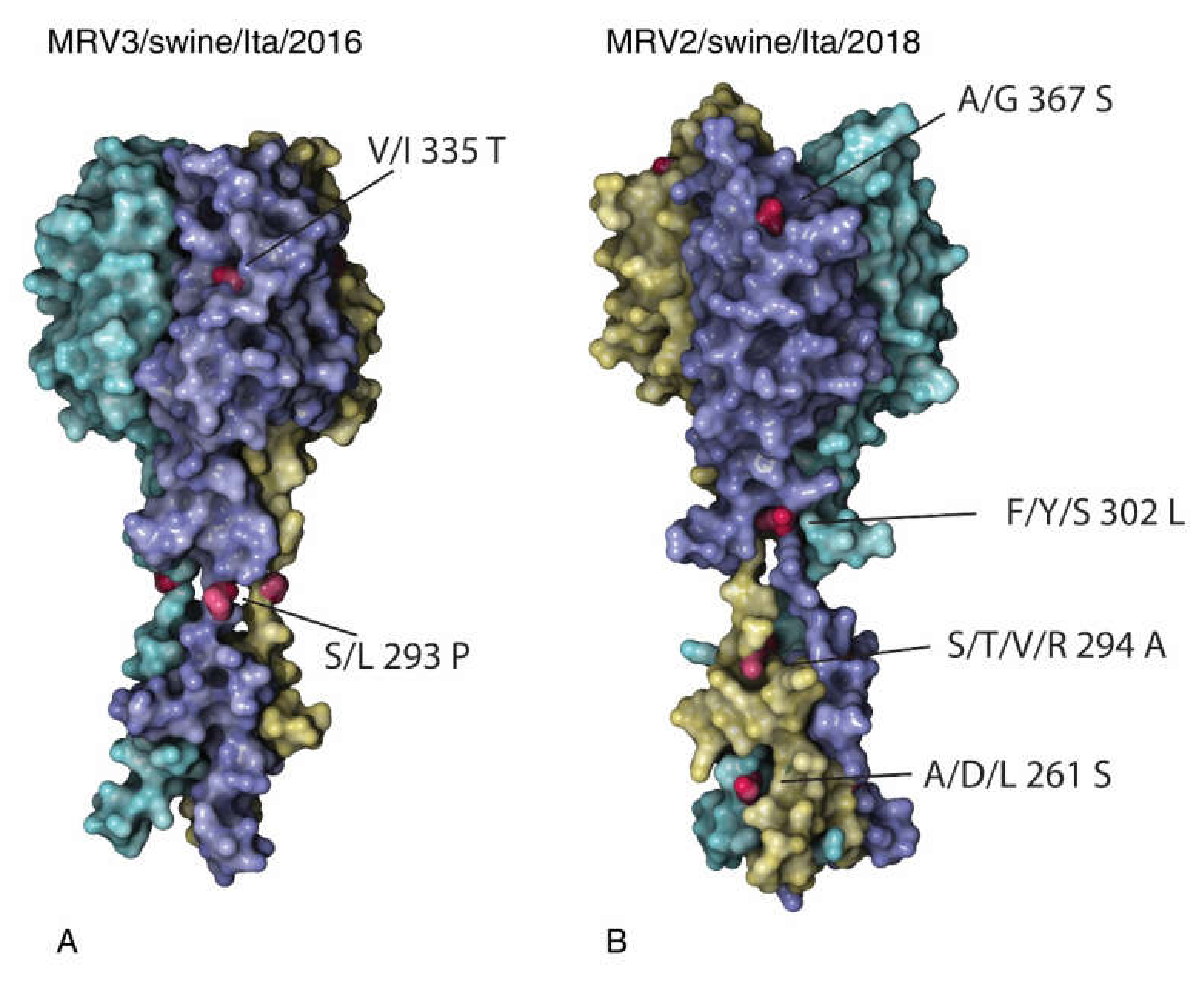
2.5. MRV2 and MRV3 S1 Homology Modelling
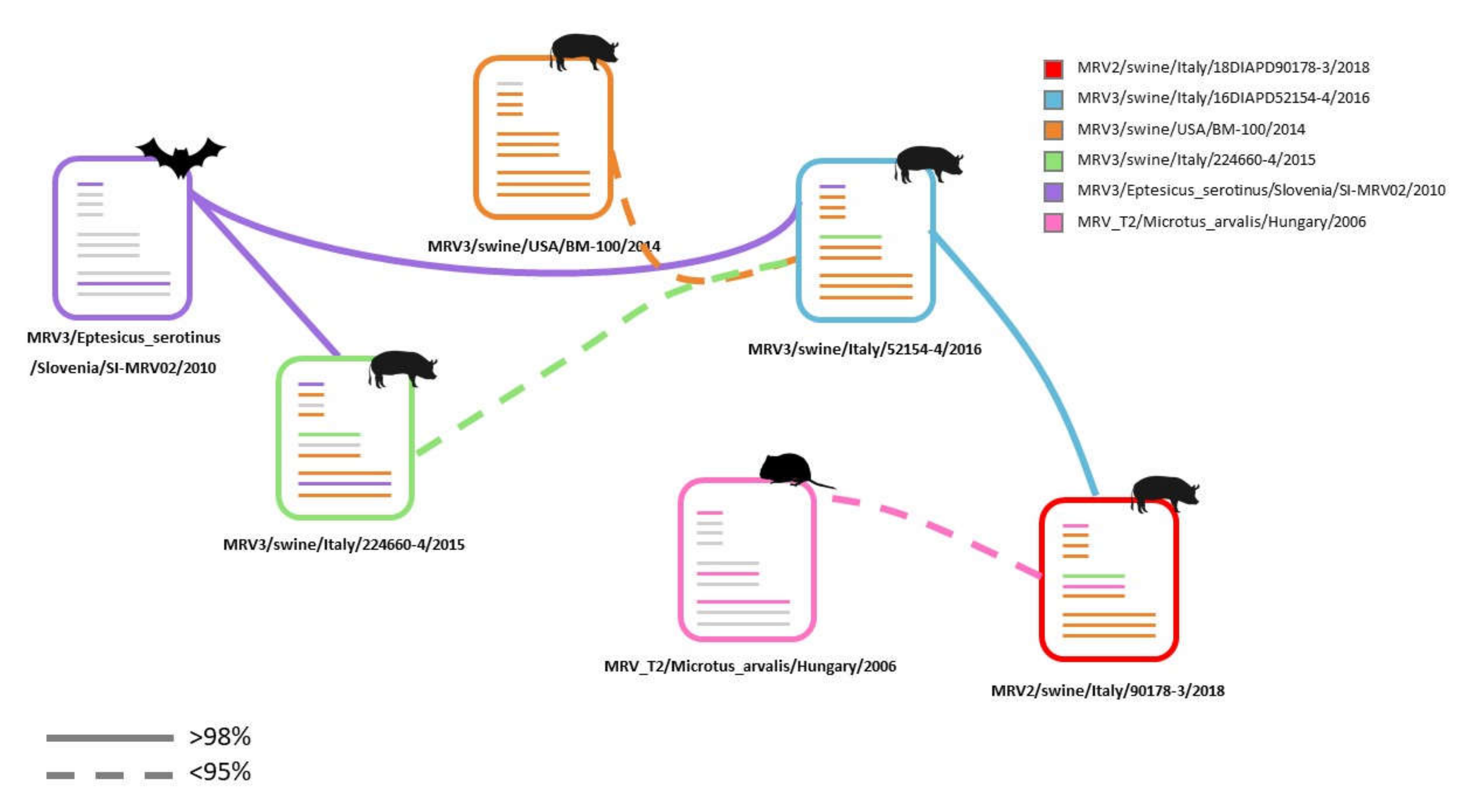
3. Results
3.1. Identification of Two Novel MRVs in One Single Swine Farm in North Eastern Italy
3.2. NGS Sequencing of Swine MRV2 and MRV3
3.3. Molecular Characterization of Swine MRV2 and MRV3
3.4. Analysis of the S1 Protein of Swine MRV3 and MRV2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Day, J.M. The Diversity of the Orthoreoviruses: Molecular Taxonomy and Phylogentic Divides. Infect. Genet. Evol. 2009, 9, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Dermody, T.S.; Parker, J.S.L.; Sherry, B. Orthoreoviruses in Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, USA, 2013; Volume 2, pp. 1304–1346. [Google Scholar]
- Ouattara, L.A.; Barin, F.; Barthez, M.A.; Bonnaud, B.; Roingeard, P.; Goudeau, A.; Castelnau, P.; Vernet, G.; Paranhos-Baccalà, G.; Komurian-Pradel, F. Novel Human Reovirus Isolated from Children with Acute Necrotizing Encephalopathy. Emerg. Infect. Dis. 2011, 17, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L. Serologic Grouping of Reoviruses by Hemagglutination-Inhibition. Am. J. Hyg. 1960, 71, 242–249. [Google Scholar] [PubMed]
- Attoui, H.; Biagini, P.; Stirling, J.; Mertens, P.P.; Cantaloube, J.F.; Meyer, A.; de Micco, P.; de Lamballerie, X. Sequence Characterization of Ndelle Virus Genome Segments 1, 5, 7, 8, and 10: Evidence for Reassignment to the Genus Orthoreovirus, Family Reoviridae. Biochem. Biophys. Res. Commun. 2001, 287, 583–588. [Google Scholar] [CrossRef]
- Attoui, H.; Mertens, P.P.C.; Becnel, J.; Belaganahalli, S.; Bergoin, M.; Brussaard, C.P.; Chappell, J.D.; Ciarlet, M.; del Vas, M.; Dermody, T.S.; et al. Reoviridae in ICTV 9th Report. 2011. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/dsrna-viruses-2011/w/dsrna_viruses/188/reoviridae (accessed on 30 March 2020).
- Rosen, L.; Hovis, J.F.; Mastrota, F.M.; Bell, J.A.; Huebner, R.J. Observations on a Newly Recognized Virus (Abney) of the Reovirus Family. Am. J. Hyg. 1960, 71, 258–265. [Google Scholar]
- Fukutomi, T.; Sanekata, T.; Akashi, H. Isolation of Reovirus Type 2 from Diarrheal Feces of Pigs. J. Vet. Med. Sci. 1996, 58, 555–557. [Google Scholar] [CrossRef]
- Dai, Y.; Zhou, Q.; Zhang, C.; Song, Y.; Tian, X.; Zhang, X.; Xue, C.; Xu, S.; Bi, Y.; Cao, Y. Complete Genome Sequence of a Porcine Orthoreovirus from Southern China. J. Virol. 2012, 86, 12456. [Google Scholar] [CrossRef]
- Fehér, E.; Kemenesi, G.; Oldal, M.; Kurucz, K.; Kugler, R.; Farkas, S.L.; Marton, S.; Horváth, G.; Bányai, K.; Jakab, F. Isolation and Complete Genome Characterization of Novel Reassortant Orthoreovirus from Common Vole (Microtus Arvalis). Virus Genes 2017, 53, 307–311. [Google Scholar] [CrossRef]
- Kohl, C.; Lesnik, R.; Brinkmann, A.; Ebinger, A.; Radonić, A.; Nitsche, A.; Mühldorfer, K.; Wibbelt, G.; Kurth, A. Isolation and Characterization of Three Mammalian Orthoreoviruses from European Bats. PLoS ONE 2012, 7, e43106. [Google Scholar] [CrossRef]
- Kwon, H.; Kim, H.; Kim, H.; Park, J.; Son, K.; Jung, J.; Lee, W.S.; Cho, K.; Park, S.; Kang, M. Detection and Molecular Characterization of Porcine Type 3 Orthoreoviruses Circulating in South Korea. Vet. Microbiol. 2012, 157, 456–463. [Google Scholar] [CrossRef]
- Lelli, D.; Beato, M.S.; Cavicchio, L.; Lavazza, A.; Chiapponi, C.; Leopardi, S.; Baioni, L.; De Benedictis, P.; Moreno, A. First Identification of Mammalian Orthoreovirus Type 3 in Diarrheic Pigs in Europe. Virol. J. 2016, 13, 139. [Google Scholar] [CrossRef]
- Lewandowska, D.W.; Capaul, R.; Prader, S.; Zagordi, O.; Geissberger, F.; Kügler, M.; Knorr, M.; Berger, C.; Güngör, T.; Reichenbach, J.; et al. Persistent Mammalian Orthoreovirus, Coxsackievirus and Adenovirus Co-Infection in a Child with a Primary Immunodeficiency Detected by Metagenomic Sequencing: A Case Report. BMC Infect. Dis. 2018, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Naglič, T.; Rihtarič, D.; Hostnik, P.; Toplak, N.; Koren, S.; Kuhar, U.; Jamnikar-Ciglenečki, U.; Kutnjak, D.; Steyer, A. Identification of Novel Reassortant Mammalian Orthoreoviruses from Bats in Slovenia. BMC Vet. Res. 2018, 14, 264. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Li, H.; Li, L.; Wang, J.; Wang, B.; Xie, R.; Xu, H.; Zhao, L.; Pan, Y.; Song, Y.; et al. Genetic and Pathogenic Characterization of a Novel Reassortant Mammalian Orthoreovirus 3 (MRV3) from a Diarrheic Piglet and Seroepidemiological Survey of MRV3 in Diarrheic Pigs from East China. Vet. Microbiol. 2017, 208, 126–136. [Google Scholar] [CrossRef]
- Thimmasandra Narayanappa, A.; Sooryanarain, H.; Deventhiran, J.; Cao, D.; Ammayappan Venkatachalam, B.; Kambiranda, D.; LeRoith, T.; Heffron, C.L.; Lindstrom, N.; Hall, K.; et al. A Novel Pathogenic Mammalian Orthoreovirus from Diarrheic Pigs and Swine Blood Meal in the United States. MBio 2015, 6, 593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, L.; Wang, P.; Liu, S.; Lin, W.; Hu, F.; Wu, W.; Chen, W.; Cui, S. A Potentially Novel Reovirus Isolated from Swine in Northeastern China in 2007. Virus Genes 2011, 43, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Mikuletič, T.; Steyer, A.; Kotar, T.; Zorec, T.M.; Poljak, M. A Novel Reassortant Mammalian Orthoreovirus with a Divergent S1 Genome Segment Identified in a Traveler with Diarrhea. Infect. Genet. Evol. 2019, 73, 378–383. [Google Scholar] [CrossRef]
- Rosa, U.A.; Ribeiro, G.O.; Villanova, F.; Luchs, A.; Milagres, F.; Komninakis, S.V.; Tahmasebi, R.; Lobato, M.; Brustulin, R.; Chagas, R.; et al. First Identification of Mammalian Orthoreovirus Type 3 by Gut Virome Analysis in Diarrheic Child in Brazil. Sci. Rep. 2019, 9, 18599. [Google Scholar] [CrossRef]
- Steyer, A.; Gutiérrez-Aguire, I.; Kolenc, M.; Koren, S.; Kutnjak, D.; Pokorn, M.; Poljšak-Prijatelj, M.; Racki, N.; Ravnikar, M.; Sagadin, M.; et al. High Similarity of Novel Orthoreovirus Detected in a Child Hospitalized with Acute Gastroenteritis to Mammalian Orthoreoviruses found in Bats in Europe. J. Clin. Microbiol. 2013, 51, 3818–3825. [Google Scholar] [CrossRef]
- Tyler, K.L.; Barton, E.S.; Ibach, M.L.; Robinson, C.; Campbell, J.A.; O’Donnell, S.M.; Valyi-Nagy, T.; Clarke, P.; Wetzel, J.D.; Dermody, T.S. Isolation and Molecular Characterization of a Novel Type 3 Reovirus from a Child with Meningitis. J. Infect. Dis. 2004, 189, 1664–1675. [Google Scholar] [CrossRef]
- Yamamoto, S.P.; Motooka, D.; Egawa, K.; Kaida, A.; Hirai, Y.; Kubo, H.; Motomura, K.; Nakamura, S.; Iritani, N. Novel Human Reovirus Isolated from Children and its Long-Term Circulation with Reassortments. Sci. Rep. 2020, 10, 963. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.C.Y.; Wang, Y.; Huang, S.; Yang, J.; Wang, J. High Incidence of Mammalian Orthoreovirus Identified by Environmental Surveillance in Taiwan. PLoS ONE 2015, 10, e0142745. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Sooryanarain, H.; Yugo, D.M.; Tian, D.; Rogers, A.J.; Heffron, C.L.; Thimmasandra Narayanappa, A.; LeRoith, T.; Overend, C.; Matzinger, S.R.; et al. Evaluation of the Pathogenicity of Mammalian Orthoreovirus Type 3 (MRV3) in Germ-Free Gnotobiotic Pigs and of the Efficacy of an Inactivated Vaccine Against MRV3 Infection in Neonatal Conventional Piglets. Vet. Microbiol. 2018, 224, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Leary, T.P.; Erker, J.C.; Chalmers, M.L.; Cruz, A.T.; Wetzel, J.D.; Desai, S.M.; Mushahwar, I.K.; Dermody, T.S. Detection of Mammalian Reovirus RNA by using Reverse Transcription-PCR: Sequence Diversity within the Lambda3-Encoding L1 Gene. J. Clin. Microbiol. 2002, 40, 1368–1375. [Google Scholar] [CrossRef]
- Chen, Q.; Li, G.; Stasko, J.; Thomas, J.T.; Stensland, W.R.; Pillatzki, A.E.; Gauger, P.C.; Schwartz, K.J.; Madson, D.; Yoon, K.; et al. Isolation and Characterization of Porcine Epidemic Diarrhea Viruses Associated with the 2013 Disease Outbreak among Swine in the United States. J. Clin. Microbiol. 2014, 52, 234–243. [Google Scholar] [CrossRef] [PubMed]
- De Carlo, S.; Harris, J.R. Negative Staining and Cryo-Negative Staining of Macromolecules and Viruses for TEM. Micron 2011, 42, 117–131. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A De Novo Assembler for Single-Cell and Metagenomic Sequencing Data with Highly Uneven Depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet--Next Generation Sequence Assembly Visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, S.; Cao, L.; Lei, W.; Cao, Y.; Song, J.; Tang, Q.; Zhang, H.; Feng, Y.; Yang, W.; et al. Isolation and Identification of a Natural Reassortant Mammalian Orthoreovirus from Least Horseshoe Bat in China. PLoS ONE 2015, 10, e0118598. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-New Features and Functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Tomasello, G.; Armenia, I.; Molla, G. The Protein Imager: A Full-Featured Online Molecular Viewer Interface with Server-Side HQ-Rendering Capabilities. Bioinformatics 2020, btaa009, 1367–4803. [Google Scholar] [CrossRef]
- Chappell, J.D.; Barton, E.S.; Smith, T.H.; Baer, G.S.; Duong, D.T.; Nibert, M.L.; Dermody, T.S. Cleavage Susceptibility of Reovirus Attachment Protein Sigma1 during Proteolytic Disassembly of Virions is Determined by a Sequence Polymorphism in the Sigma1 Neck. J. Virol. 1998, 72, 8205–8213. [Google Scholar] [CrossRef]
- Bassel-Duby, R.; Spriggs, D.R.; Tyler, K.L.; Fields, B.N. Identification of Attenuating Mutations on the Reovirus Type 3 S1 Double-Stranded RNA Segment with a Rapid Sequencing Technique. J. Virol. 1986, 60, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.D.; Gunn, V.L.; Wetzel, J.D.; Baer, G.S.; Dermody, T.S. Mutations in Type 3 Reovirus that Determine Binding to Sialic Acid are Contained in the Fibrous Tail Domain of Viral Attachment Protein Sigma1. J. Virol. 1997, 71, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tan, B.; Wang, B.; Li, W.; Wang, N.; Luo, C.; Wang, M.; Zhang, W.; Li, B.; Peng, C.; et al. Isolation and Identification of Bat Viruses Closely Related to Human, Porcine and Mink Orthoreoviruses. J. Gen. Virol. 2015, 96, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Lelli, D.; Moreno, A.; Steyer, A.; Nagliˇc, T.; Chiapponi, C.; Prosperi, A.; Faccin, F.; Sozzi, E.; Lavazza, A. Detection and Characterization of a Novel Reassortant Mammalian Orthoreovirus in Bats in Europe. Viruses 2015, 7, 5844–5854. [Google Scholar] [CrossRef]
- Li, Z.; Liu, D.; Ran, X.; Liu, C.; Guo, D.; Hu, X.; Tian, J.; Zhang, X.; Shao, Y.; Liu, S.; et al. Characterization and Pathogenicity of a Novel Mammalian Orthoreovirus from Wild Short-Nosed Fruit Bats. Infect. Genet. Evol. 2016, 43, 347–353. [Google Scholar] [CrossRef]
- Betancourt, W.Q.; Gerba, C.P. Rethinking the Significance of Reovirus in Water and Wastewater. Food Environ. Virol. 2016, 8, 161–173. [Google Scholar] [CrossRef]
- Chua, K.B.; Voon, K.; Yu, M.; Keniscope, C.; Abdul Rasid, K.; Wang, L. Investigation of a Potential Zoonotic Transmission of Orthoreovirus Associated with Acute Influenza-Like Illness in an Adult Patient. PLoS ONE 2011, 6, e25434. [Google Scholar] [CrossRef]
- Jiang, J.; Hermann, L.; Coombs, K.M. Genetic Characterization of a New Mammalian Reovirus, Type 2 Winnipeg (T2W). Virus Genes 2006, 33, 193–204. [Google Scholar] [CrossRef]
- Li, Z.; Shao, Y.; Liu, C.; Liu, D.; Guo, D.; Qiu, Z.; Tian, J.; Zhang, X.; Liu, S.; Qu, L. Isolation and Pathogenicity of the Mammalian Orthoreovirus MPC/04 from Masked Civet Cats. Infect. Genet. Evol. 2015, 36, 55–61. [Google Scholar] [CrossRef]
- Lelli, D.; Moreno, A.; Lavazza, A.; Bresaola, M.; Canelli, E.; Boniotti, M.B.; Cordioli, P. Identification of Mammalian Orthoreovirus Type 3 in Italian Bats. Zoonoses Public Health 2013, 60, 84–92. [Google Scholar] [CrossRef]
- Hu, T.; Qiu, W.; He, B.; Zhang, Y.; Yu, J.; Liang, X.; Zhang, W.; Chen, G.; Zhang, Y.; Wang, Y.; et al. Characterization of a Novel Orthoreovirus Isolated from Fruit Bat, China. BMC Microbiol. 2014, 14, 293. [Google Scholar] [CrossRef] [PubMed]
- Lanoie, D.; Côté, S.; Degeorges, E.; Lemay, G. A Single Mutation in the Mammalian Orthoreovirus S1 Gene is Responsible for Increased Interferon Sensitivity in a Virus Mutant Selected in Vero Cells. Virology 2019, 528, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Sandekian, V.; Lemay, G. A Single Amino Acid Substitution in the mRNA Capping Enzyme Λ2 of a Mammalian Orthoreovirus Mutant Increases Interferon Sensitivity. Virology 2015, 483, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Sandekian, V.; Lemay, G. Amino Acids Substitutions in Σ1 and Μ1 Outer Capsid Proteins of a Vero Cell-Adapted Mammalian Orthoreovirus are Required for Optimal Virus Binding and Disassembly. Virus Res. 2015, 196, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Lowen, A.C. It’s in the Mix: Reassortment of Segmented Viral Genomes. PLoS Pathog. 2018, 14, e1007200. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MRV3/Swine/Italy/52154-4/2016 | MRV2/Swine/Italy/90178-3/2018 | |||||||
|---|---|---|---|---|---|---|---|---|
| % nt | % aa | % nt | % aa | |||||
| L1 | MRV2/swine/Italy/90178-3/2018 | 99.57 | MRV/RUS/Moscow/2017 | 100 | MRV3/swine/Italy/52154-4/2016 | 99.57 | MRV3/swine/Italy/52154-4/2016 | 99.91 |
| MRV3/swine/Italy/224660-4/2015 | 94.90 | MRV3/swine/Italy/90178-3/2018 | 99.91 | MRV3/swine/Italy/224660-4/2015 | 94.93 | MRV3/swine/Italy/224660-4/2015 | 98.89 | |
| MRV_T3_Dearing_Vero_adapted_virus/VeroAV | 92.15 | MRV3/swine/Italy/224660-4/2015 | 98.95 | MRV_T3_Dearing_Vero_adapted_virus/VeroAV | 92.01 | MRV3/swine/USA/FS-03/2014 | 98.66 | |
| L2 | MRV2_swine/Italy/90178-3/2018 | 99.40 | MRV2_swine/Italy/D1690178-3/2018 | 99.55 | MRV3/swine/Italy/52154-4/2016 | 99.40 | MRV3/swine/Italy/52154-4/2016 | 99.55 |
| MRV_T3/murine/France/1961 | 90.42 | MRV_T3/murine/France/1961 | 97.39 | MRV_T3/murine/France/1961 | 90.21 | MRV_T3/murine/France/1961 | 97.51 | |
| MRV_T1/human/Ohio/1953 | 89.61 | MRV_T1/human/Ohio/1953 | 96.94 | MRV_T1/human/Ohio/1953 | 89.60 | MRV_T1/human/Ohio/1953 | 97.11 | |
| L3 | MRV2_swine/Italy/90178-3/2018 | 99.69 | MRV2/swine/Italy/90178-3/2018 | 99.90 | MRV3/swine/Italy/52154-4/2016 | 99.69 | MRV3/swine/Italy/52154-4/2016 | 99.90 |
| MRV3/swine/Italy/224660-4/2015 | 94.62 | MRV3/swine/USA/FS-03/2014 | 99.72 | MRV3/swine/Italy/224660-4/2015 | 94.31 | MRV1/swine/China/SHR-A/2011 | 98.81 | |
| MRV1_Lang | 93.23 | MRV1/swine/China/SHR-A/2011 | 99.71 | MRV1_Lang | 92.71 | MRV3_chamois/Italy/84407/2009 | 98.66 | |
| M1 | MRV2_swine/Italy/90178-3/2018 | 99.24 | MRV2/swine/Italy/90178-3/2018 | 99.01 | MRV3/swine/Italy/52154-4/2016 | 99.24 | MRV3/swine/Italy/52154-4/2016 | 99.00 |
| MRV2/human/MRV2Tou05/France | 91.86 | MRV2/human/MRV2Tou05/France | 95.31 | MRV2/human/MRV2Tou05/France | 92.27 | MRV2/human/MRV2Tou05/France | 96.06 | |
| MRV1/swine/China/SHR-A/2011 | 89.40 | MRV1_T1/human/Ohio/1953 | 94.74 | MRV1/swine/China/SHR-A/2011 | 89.73 | MRV1_T1/human/Ohio/1953 | 95.38 | |
| M2 | MRV_T1/human/Ohio/1953 | 92.57 | MRV_T1/human/Ohio/1953 | 98.45 | MRV_T2/microtus_arvalis/Hungary/2006 | 90.65 | MRV3/tree_shrew/China/2012 | 96.57 |
| MRV1_b | 92.52 | MRV1_b | 98.30 | MRV3_human/Abney | 89.99 | MRV_T2/microtus_arvalis/Hungary/2006 | 96.57 | |
| MRV1 | 92.20 | MRV1 | 97.88 | MRV1/swine/China/SHR-A/2011 | 89.90 | MRV1/tree_shrew/China/2011 | 96.42 | |
| M3 | MRV2/swine/Italy/90178-3/2018 | 98.57 | MRV3/swine/Italy/224660-4/2015 | 96.98 | MRV3/swine/Italy/52154-4/2016 | 98.57 | MRV3/swine/Italy/52154-4/2016 | 99.20 |
| MRV3/swine/Italy/224660-4/2015 | 92.11 | MRV3/swine/USA/BM-100/2014 | 96.02 | MRV3/swine/Italy/224660-4/2015 | 91.83 | MRV3/swine/Italy/224660-4/2015 | 97.09 | |
| MRV3/swine/USA/BM-100/2014 | 90.26 | MRV3/swine/USA/FS-03/2014 | 96.02 | MRV3/swine/USA/BM-100/2014 | 89.75 | MRV3/swine/USA/BM-100/2014 | 96.25 | |
| S1 | MRV3/eptesicus_serotinus/Slovenia/SI-MRV02/2010 | 98.37 | MRV3/eptesicus_serotinus/Slovenia/SI-MRV02/2010 | 98.45 | MRV2/swine/Taiwan/sR1590/2015 | 88.55 | MRV2/swine/Taiwan/sR1590/2015 | 88.91 |
| T3/pipistrellus_Khulii/Italy/5515-2/2012 | 98.23 | T3/pipistrellus_Khulii/Italy/5515-2/2012 | 98.45 | MRV_T2/microtus_arvalis/Hungary/2006 | 85.19 | MRV2/myotis_myotis/Slovenia/SI-MRV05/2008 | 87.80 | |
| MRV/human/Slovenia/SI-MRV01/2013 | 98.15 | MRV/human/Slovenia/SI-MRV01/2013 | 98.23 | MRV2/myotis_myotis/Slovenia/SI-MRV05/2008 | 84.89 | MRV_T2/microtus_arvalis/Hungary/2006 | 87.58 | |
| S2 | MRV2/swine/Italy/90178-3/2018 | 98.86 | MRV2/swine/Italy/90178-3/2018 | 99.51 | MRV3/swine/Italy/52154-4/2016 | 98.86 | MRV3/swine/Italy/52154-4/2016 | 99.51 |
| MRV2/swine/Taiwan/sR1590/2015 | 93.18 | MRV2/odocoileus virginianus/USA/OV204/2016 | 99.02 | MRV_1_Lang_Prototype | 93.24 | MRV2/odocoileus virginianus/USA/OV204/2016 | 99.04 | |
| MRV2/swine/Taiwan/sR1677/2015 | 92.53 | MRV1/tree shrew/China/2011 | 99.02 | MRV2/swine/Taiwan/sR1590/2015 | 92.76 | MRV1/tree shrew/China/2011 | 99.04 | |
| S3 | MRV2/swine/Italy/90178-3/2018 | 99.52 | MRV2/swine/Italy/90178-3/2018 | 99.41 | MRV3/swine/Italy/52154-4/2016 | 99.52 | MRV3/swine/Italy/52154-4/2016 | 99.41 |
| T3/bovine/Maryland/clone18/1961 | 94.00 | MRV3/human/Brazil/AP-151/2015 | 98.55 | T3/bovine/Maryland/clone18/1961 | 94.37 | MRV3/swine/USA/FS-03/2014 | 98.90 | |
| MRV3/swine/USA/FS-03/2014 | 93.71 | MRV3/swine/USA/FS-03/2014 | 98.25 | T1/human/Wash.D.C/clone62/1957 | 94.10 | MRV3/human/Brazil/AP-151/2015 | 98.66 | |
| S4 | MRV3/swine/China/GD-1/2012 | 94.80 | MRV2/swine/Italy/90178-3/2018 | 98.09 | MRV1/swine/China/SHR-A/2011 | 94.44 | MRV3/swine/Italy/52154-4/2016 | 98.09 |
| MRV1/swine/China/SHR-A/2011 | 94.37 | MRV1/swine/China/SHR-A/2011 | 97.77 | MRV3/swine/Italy/224660-4/2015 | 94.08 | MRV1/swine/China/SHR-A/2011 | 98.08 | |
| MRV3/swine/Italy/224660-4/2015 | 93.63 | MRV3/swine/China/GD-1/2012 | 97.45 | MRV3/swine/Italy/52154-4/2016 | 93.52 | MRV3/swine/Italy/224660-4/2015 | 97.26 | |
| Nucleotide Positions | 37 39 | 73 75 | 76 78 | 112 114 | 331 333 | 346 348 | 388 390 | 406 408 | 460 462 | 472 474 | 514 516 | 649 651 | 739 741 | 799 801 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino acid position | 13 | 25 | 26 | 38 | 111 | 116 | 130 | 136 | 154 | 158 | 172 | 217 | 247 | 267 |
| MRV2/swine/Italy/18DIAPD90178-3/2018 | F | G | L | L | V | L | G | S | L | Q | L | A | G | S |
| MRV_T2/microtus_arvalis/Hungary/2006 | F | G | L | L | V | L | G | S | L | Q | L | A | G | S |
| MRV2/myotis_myotis/Slovenia/SI-MRV05/2008 | F | G | L | L | V | L | G | S | L | Q | L | A | G | S |
| MRV2/swine/Taiwan/sR1590/2015 | F | G | L | L | V | L | G | S | L | Q | L | A | G | S |
| MRV2/swine/Taiwan/sR1677/2015 | L | E | I | N | A | V | N | N | S | G | T | S | S | N |
| MRV2/swine/Taiwan/sR1521/2015 | L | E | I | N | A | V | N | N | S | G | T | S | S | N |
| MRV2/human/Japan/Osaka2014/2014 | L | E | I | T | T | I | N | T | A | S | T | S | S | N |
| MRV2/human/Japan/Osaka2005/2005 | L | E | I | T | T | I | N | T | A | S | T | S | S | N |
| MRV2/human/Japan/Osaka1994/1994 | L | E | I | T | T | I | N | T | A | S | T | S | S | N |
| MRV2/hipposideros/China/WIV5/2011 | L | E | I | N | T | I | N | T | A | S | T | S | S | N |
| MRV2/hipposideros/China/WIV3/2011 | L | E | I | N | A | V | N | T | G | G | T | S | S | N |
| MRV/horseshoe_bat/China/RpMRV- YN2012/2012 | L | E | I | N | A | V | N | N | N | G | T | S | S | N |
| MRV2/Nanzi9A/Taiwan/2013 | - | - | - | T | T | I | N | T | V | S | T | S | S | N |
| MRV2/Taiwan/2013 (10 strains) | - | - | - | T | T | I | N | T | A | S | T | S | S | N |
| MRV2/panthera_leo/Japan/2011 | L | E | I | N | T | V | N | N | N | G | T | S | S | N |
| MRV2/odocoileus_virginianus/USA/OV204/2016 | L | E | I | N | T | V | N | N | S | G | T | S | S | N |
| MRV2/hipposideros/China/WIV4/2011 | L | E | I | N | T | V | K | N | G | D | T | S | S | N |
| MRV2/human/MRV2Tou05/France | L | E | T | N | T | V | N | T | G | G | T | S | S | N |
| MRV2/myodes_glareolus/Germany/TRALAU2004/2004 | L | E | I | N | A | I | R | N | D | D | T | - | S | N |
| MRV2_302I/human/China/1982 | L | E | I | N | A | V | N | N | S | G | T | S | S | N |
| Most common amino acid in NCBI available sequences | L | E | I | N | T | I | N | T | A | S | T | S | S | N |
| Other amino acid variants in NCBI available sequences | I | - | V,T | T,A,S | A,S,G | V,R,T | T, E, K, D, R, S | N, A | S, Q, G, N, V, D | G, A, V, D | - | N | A | Q |
| Nucleotide Position * | Amino Acid Position | Unique Amino Acid Mutations of MRV2/Swine/Ita/2018 | Typical Amino Acids of MRV2 Strains |
|---|---|---|---|
| 1–3 | 1 | L | M |
| 226–228 | 76 | V | T,L,A |
| 241–243 | 81 | I | S,A |
| 355–357 | 119 | S | D,N,V |
| 361–363 | 121 | V | S,T,A,L,I |
| 478–480 | 160 | G | V,D,N,S,A |
| 571–573 | 191 | G | N,T |
| 589–591 | 197 | D | N,S,G,A,R |
| 640–642 | 214 | L | F |
| 655–657 | 219 | V | M,I,L |
| 781–783 | 261 | S | A,D,L |
| 880–882 | 294 | A | S,T,V,R |
| 904–906 | 302 | L | F,Y,S |
| 1099–1101 | 367 | S | G,A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavicchio, L.; Tassoni, L.; Zamperin, G.; Campalto, M.; Carrino, M.; Leopardi, S.; De Benedictis, P.; Beato, M.S. Unexpected Genetic Diversity of Two Novel Swine MRVs in Italy. Viruses 2020, 12, 574. https://doi.org/10.3390/v12050574
Cavicchio L, Tassoni L, Zamperin G, Campalto M, Carrino M, Leopardi S, De Benedictis P, Beato MS. Unexpected Genetic Diversity of Two Novel Swine MRVs in Italy. Viruses. 2020; 12(5):574. https://doi.org/10.3390/v12050574
Chicago/Turabian StyleCavicchio, Lara, Luca Tassoni, Gianpiero Zamperin, Mery Campalto, Marilena Carrino, Stefania Leopardi, Paola De Benedictis, and Maria Serena Beato. 2020. "Unexpected Genetic Diversity of Two Novel Swine MRVs in Italy" Viruses 12, no. 5: 574. https://doi.org/10.3390/v12050574
APA StyleCavicchio, L., Tassoni, L., Zamperin, G., Campalto, M., Carrino, M., Leopardi, S., De Benedictis, P., & Beato, M. S. (2020). Unexpected Genetic Diversity of Two Novel Swine MRVs in Italy. Viruses, 12(5), 574. https://doi.org/10.3390/v12050574