Global Transmission, Spatial Segregation, and Recombination Determine the Long-Term Evolution and Epidemiology of Bovine Coronaviruses

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Sample Preparation and NGS Sequencing
2.3. Screening for Bovine Respiratory Pathogens
2.4. BCoV Isolation
2.5. Virus sequence Datasets and Phylogenetic Analysis
2.6. Recombination Detection
2.7. Molecular Dating
2.8. Selective Pressure Analysis
3. Results
3.1. Genomic Surveillance of BCoV in France
3.2. Recombination of BCoV
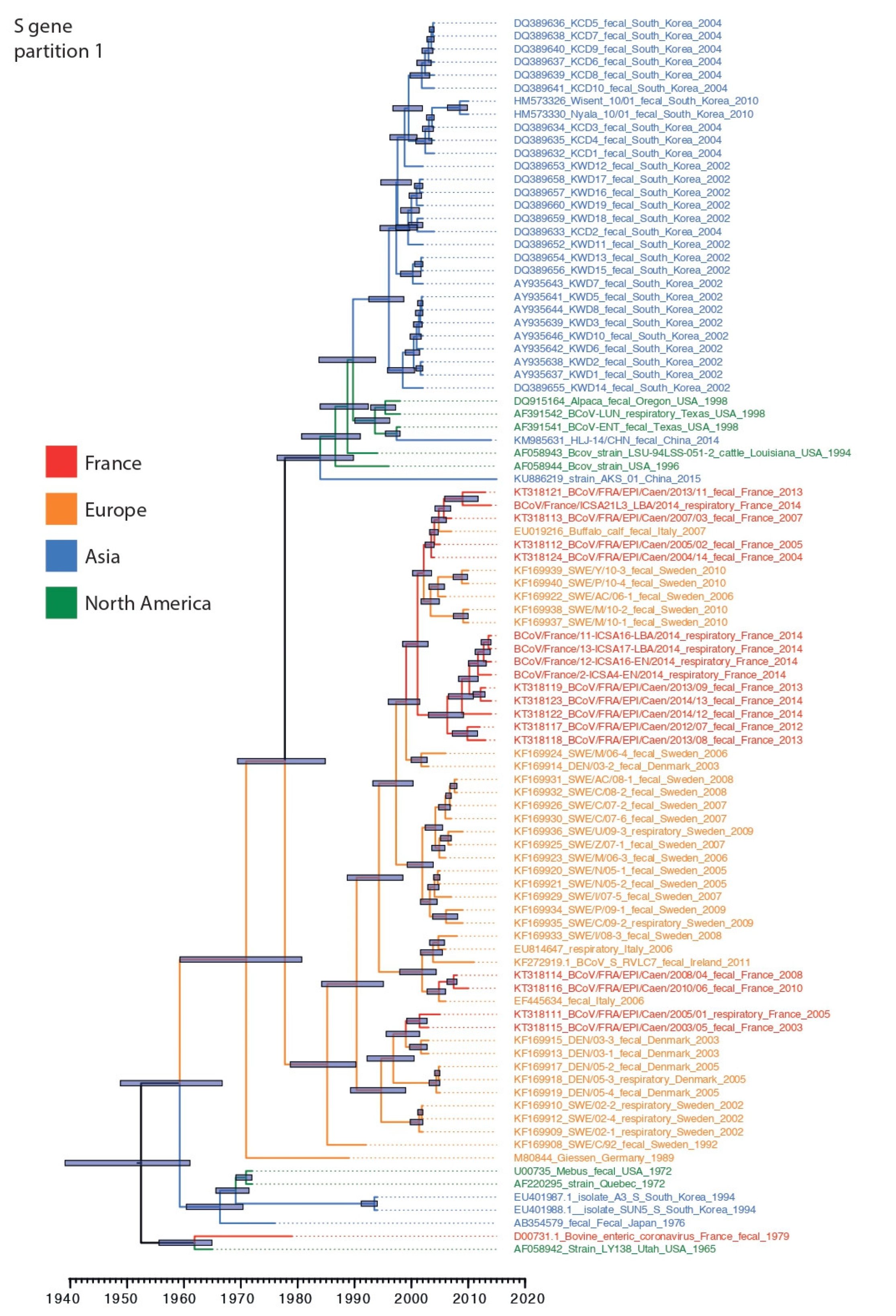
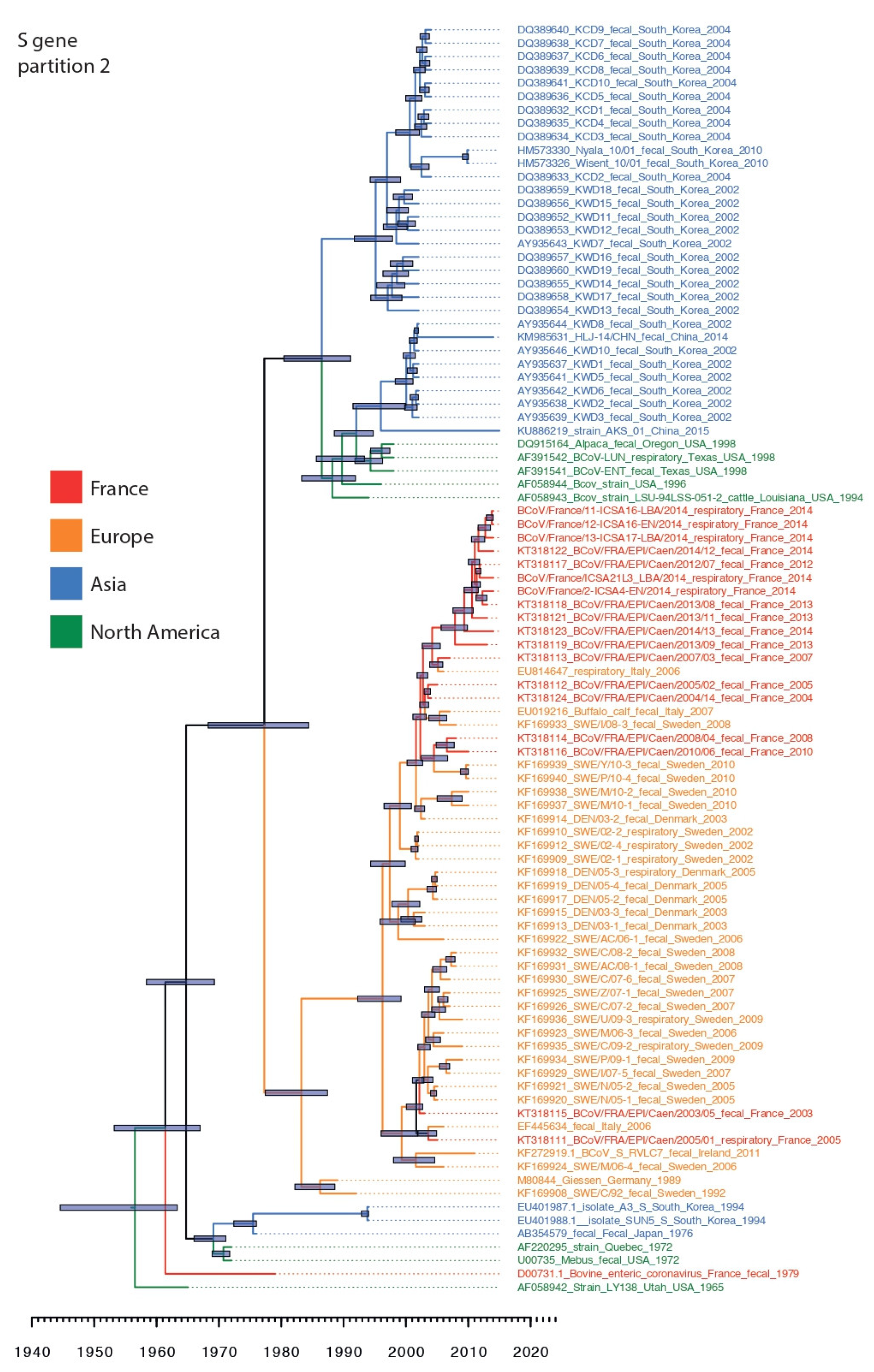
3.3. Phylogenetic Relationships and Spatio-Temporal Evolution of BCoV
3.4. Selection Pressure
4. Discussion
4.1. Prevalence of BCoV in Europe
4.2. No obvious Link between Virus Tropism and Genetic Markers
4.3. Little Exchanges of BCoV between Europe and the Other Continents
4.4. Evolution of BCOV
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grissett, G.P.; White, B.J.; Larson, R.L. Structured Literature Review of Responses of Cattle to Viral and Bacterial Pathogens Causing Bovine Respiratory Disease Complex. J. Vet. Intern. Med. 2015, 29, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Caswell, J.L. Failure of respiratory defenses in the pathogenesis of bacterial pneumonia of cattle. Vet. Pathol. 2014, 51, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Hilton, W.M. BRD in 2014: Where have we been, where are we now, and where do we want to go? Anim. Health Res. Rev. 2014, 15, 120–122. [Google Scholar] [CrossRef]
- Hodgson, P.D.; Aich, P.; Stookey, J.; Popowych, Y.; Potter, A.; Babiuk, L.; Griebel, P.J. Stress significantly increases mortality following a secondary bacterial respiratory infection. Vet. Res. 2012, 43, 21. [Google Scholar] [CrossRef]
- Hodgson, P.D.; Aich, P.; Manuja, A.; Hokamp, K.; Roche, F.M.; Brinkman, F.S.L.; Potter, A.; Babiuk, L.A.; Griebel, P.J. Effect of stress on viral-bacterial synergy in bovine respiratory disease: Novel mechanisms to regulate inflammation. Comp. Funct. Genomics 2005, 6, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Risalde, M.A.; Molina, V.; Sánchez-Cordón, P.J.; Romero-Palomo, F.; Pedrera, M.; Garfia, B.; Gómez-Villamandos, J.C. Pathogenic mechanisms implicated in the intravascular coagulation in the lungs of BVDV-infected calves challenged with BHV-1. Vet. Res. 2013, 44, 1–13. [Google Scholar] [CrossRef]
- Valarcher, J.-F.; Taylor, G. Bovine respiratory syncytial virus infection. Vet. Res. 2007, 38, 153–180. [Google Scholar] [CrossRef]
- Boukahil, I.; Czuprynski, C.J. Mannheimia haemolytica biofilm formation on bovine respiratory epithelial cells. Vet. Microbiol. 2016, 197, 129–136. [Google Scholar] [CrossRef]
- Elswaifi, S.F.; Scarratt, W.K.; Inzana, T.J. The role of lipooligosaccharide phosphorylcholine in colonization and pathogenesis of Histophilus somni in cattle. Vet. Res. 2012, 43. [Google Scholar] [CrossRef]
- Sandal, I.; Shao, J.Q.; Annadata, S.; Apicella, M.A.; Boye, M.; Jensen, T.K.; Saunders, G.K.; Inzana, T.J. Histophilus somni biofilm formation in cardiopulmonary tissue of the bovine host following respiratory challenge. Microbes Infect. 2009, 11, 254–263. [Google Scholar] [CrossRef]
- Zecchinon, L.; Fett, T.; Desmecht, D. How Mannheimia haemolytica defeats host defence through a kiss of death mechanism. Vet. Res. 2005, 36, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.E.; Arroyo, L.G.; Poljak, Z.; Viel, L.; Weese, J.S. Detection of Bovine Coronavirus in Healthy and Diarrheic Dairy Calves. J. Vet. Intern. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.; Lorenzetti, E.; Alfieri, A.F.; Alfieri, A.A. Molecular detection of bovine coronavirus in a diarrhea outbreak in pasture-feeding Nellore steers in southern Brazil. Trop. Anim. Health Prod. 2016, 48, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Alkan, F.; Ozkul, A.; Bilge-Dagalp, S.; Karaoglu, T.; Oguzoglu, T.C.; Caliskan, E.; Burgu, I. The Detection and Genetic Characterization Based on the S1 Gene Region of BCoVs from Respiratory and Enteric Infections in Turkey. Transbound. Emerg. Dis. 2011, 58, 179–185. [Google Scholar] [CrossRef]
- Hasoksuz, M.; Hoet, A.E.; Loerch, S.C.; Wittum, T.E.; Nielsen, P.R.; Saif, L.J. Detection of respiratory and enteric shedding of bovine coronaviruses in cattle in an Ohio feedlot. J. Vet. Diagn. Investig. 2002, 14, 308–313. [Google Scholar] [CrossRef]
- Fulton, R.W.; Step, D.L.; Wahrmund, J.; Burge, L.J.; Payton, M.E.; Cook, B.J.; Burken, D.; Richards, C.J.; Confer, A.W. Bovine coronavirus (BCV) infections in transported commingled beef cattle and sole-source ranch calves. Can. J. Vet. Res. 2011, 75, 191–199. [Google Scholar]
- Fulton, R.W.; Ridpath, J.F.; Burge, L.J. Bovine coronaviruses from the respiratory tract: Antigenic and genetic diversity. Vaccine 2013, 31, 886–892. [Google Scholar] [CrossRef]
- Saif, L.J. Bovine respiratory coronavirus. Vet. Clin. North Am.—Food Anim. Pract. 2010, 26, 349–364. [Google Scholar] [CrossRef]
- Decaro, N.; Campolo, M.; Desario, C.; Cirone, F.; D’Abramo, M.; Lorusso, E.; Greco, G.; Mari, V.; Colaianni, M.L.; Elia, G.; et al. Respiratory disease associated with bovine coronavirus infection in cattle herds in Southern Italy. J. Vet. Diagnostic Investig. 2008, 20, 28–32. [Google Scholar] [CrossRef]
- Bok, M.; Miño, S.; Rodriguez, D.; Badaracco, A.; Nuñes, I.; Souza, S.P.; Bilbao, G.; Louge Uriarte, E.; Galarza, R.; Vega, C.; et al. Molecular and antigenic characterization of bovine Coronavirus circulating in Argentinean cattle during 1994-2010. Vet. Microbiol. 2015, 181, 221–229. [Google Scholar] [CrossRef]
- Toftaker, I.; Sanchez, J.; Stokstad, M.; Nødtvedt, A. Bovine respiratory syncytial virus and bovine coronavirus antibodies in bulk tank milk – risk factors and spatial analysis. Prev. Vet. Med. 2016, 133, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, K.P.; Vlasova, A.N.; Jung, K.; Hasoksuz, M.; Zhang, X.; Halpin, R.; Wang, S.; Ghedin, E.; Spiro, D.; Saif, L.J. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. J. Virol. 2008, 82, 12422–12431. [Google Scholar] [CrossRef] [PubMed]
- Vilček, S.; Jacková, A.; Kolesárová, M.; Vlasáková, M. Genetic variability of the S1 subunit of enteric and respiratory bovine coronavirus isolates. Acta Virol. 2017, 61, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Hasoksuz, M.; Lathrop, S.; Al-dubaib, M.A.; Lewis, P.; Saif, L.J. Antigenic variation among bovine enteric coronaviruses (BECV) and bovine respiratory coronaviruses (BRCV) detected using monoclonal antibodies. Arch. Virol. 1999, 144, 2441–2447. [Google Scholar] [CrossRef]
- Gélinas, A.M.; Boutin, M.; Sasseville, A.M.J.; Dea, S. Bovine coronaviruses associated with enteric and respiratory diseases in Canadian dairy cattle display different reactivities to anti-HE monoclonal antibodies and distinct amino acid changes in their HE, S and ns4.9 protein. Virus Res. 2001, 76, 43–57. [Google Scholar] [CrossRef]
- Zhang, X.; Hasoksuz, M.; Spiro, D.; Halpin, R.; Wang, S.; Vlasova, A.; Janies, D.; Jones, L.R.; Ghedin, E.; Saif, L.J. Quasispecies of bovine enteric and respiratory coronaviruses based on complete genome sequences and genetic changes after tissue culture adaptation. Virology 2007, 363, 1–10. [Google Scholar] [CrossRef]
- Gunn, L.; Collins, P.J.; O’Connell, M.J.; O’Shea, H. Phylogenetic investigation of enteric bovine coronavirus in Ireland reveals partitioning between European and global strains. Ir. Vet. J. 2015, 68, 31. [Google Scholar] [CrossRef]
- Kin, N.; Miszczak, F.; Diancourt, L.; Caro, V.; Moutou, F.; Vabret, A.; Ar Gouilh, M. Comparative molecular epidemiology of two closely related coronaviruses, bovine coronavirus (BCoV) and human coronavirus OC43 (HCoV-OC43), reveals a different evolutionary pattern. Infect. Genet. Evol. 2016, 40, 186–191. [Google Scholar] [CrossRef]
- Suzuki, T.; Otake, Y.; Uchimoto, S.; Hasebe, A.; Goto, Y. Genomic characterization and phylogenetic classification of bovine coronaviruses through whole genome sequence analysis. Viruses 2020, 12, 183. [Google Scholar] [CrossRef]
- Kanno, T.; Hatama, S.; Ishihara, R.; Uchida, I. Molecular analysis of the S glycoprotein gene of bovine coronaviruses isolated in Japan from 1999 to 2006. J. Gen. Virol. 2007, 88, 1218–1224. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Lemey, P.; Moës, E.; Li, S.; Vandamme, A.M.; Van Ranst, M. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virology 2005, 337, 85–92. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fields, M.N.; Masters, P.; Perlman, S. Coronaviridae. In Fields Virology, 6th ed.; Knipe, D.M., Ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 825–857. ISBN 9781451105636. [Google Scholar]
- Wertheim, J.O.; Chu, D.K.W.; Peiris, J.S.M.; Kosakovsky Pond, S.L.; Poon, L.L.M. A Case for the Ancient Origin of Coronaviruses. J. Virol. 2013, 87, 7039–7045. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Coronaviruses 2015, 1282, 1–23. [Google Scholar]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Gralinski, L.E.; Menachery, V.D. Return of the coronavirus: 2019-nCoV. Viruses 2020, 12, 135. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 89, 44–48. [Google Scholar] [CrossRef]
- Nemoto, M.; Kanno, T.; Bannai, H.; Tsujimura, K.; Yamanaka, T.; Kokado, H. Antibody response to equine coronavirus in horses inoculated with a bovine coronavirus vaccine. J. Vet. Med. Sci. 2017, 79, 1889–1891. [Google Scholar] [CrossRef]
- Decaro, N.; Buonavoglia, C. Canine Coronavirus: Not Only an Enteric Pathogen. Vet. Clin. North Am.—Small Anim. Pract. 2011, 41, 1121–1132. [Google Scholar] [CrossRef]
- Dong, B.; Lu, H.; Zhao, K.; Liu, W.; Gao, W.; Lan, Y.; Zhao, J.; Tang, B.; Song, D.; He, W.; et al. Identification and genetic characterization of porcine hemagglutinating encephalomyelitis virus from domestic piglets in China. Arch. Virol. 2014, 159, 2329–2337. [Google Scholar] [CrossRef]
- Van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 2012, 3, e00473-12. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Li, K.S.M.; Tsang, A.K.L.; Lam, C.S.F.; Ahmed, S.; Chen, H.; Chan, K.-H.; Woo, P.C.Y.; Yuen, K.-Y. Genetic Characterization of Betacoronavirus Lineage C Viruses in Bats Reveals Marked Sequence Divergence in the Spike Protein of Pipistrellus Bat Coronavirus HKU5 in Japanese Pipistrelle: Implications for the Origin of the Novel Middle East Respiratory Sy. J. Virol. 2013, 87, 8638–8650. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Du, L.; Liu, C.; Wang, L.; Ma, C.; Tang, J.; Baric, R.S.; Jiang, S.; Li, F. Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat-to-human transmission of MERS coronavirus. Proc. Natl. Acad. Sci. USA 2014, 111, 12516–12521. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Poon, R.W.S.; Wong, B.H.L.; Wang, M.; Huang, Y.; Xu, H.; Guo, R.; Li, K.S.M.; Gao, K.; Chan, K.-H.; et al. Coexistence of Different Genotypes in the Same Bat and Serological Characterization of Rousettus Bat Coronavirus HKU9 Belonging to a Novel Betacoronavirus Subgroup. J. Virol. 2010, 84, 11385–11394. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Wang, Q.; Qi, J.; Yuan, Y.; Xuan, Y.; Han, P.; Wan, Y.; Ji, W.; Li, Y.; Wu, Y.; Wang, J.; et al. Bat origins of MERS-CoV supported by bat Coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 2014, 16, 328–337. [Google Scholar] [CrossRef]
- Han, H.-J.; Yu, H.; Yu, X.-J. Evidence for zoonotic origins of Middle East respiratory syndrome coronavirus. J. Gen. Virol. 2016, 97, 274–280. [Google Scholar] [CrossRef]
- Mohd, H.A.; Al-Tawfiq, J.A.; Memish, Z.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) origin and animal reservoir. Virol. J. 2016, 13, 87. [Google Scholar] [CrossRef]
- Lau, S.; Wong, A.; Lau, T.; Woo, P. Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir? Int. J. Mol. Sci. 2017, 18, 2138. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2017, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Lemey, P.; Vandamme, A.; Ranst, V.; Moe, E.; Wollants, E.; Ranst, M.V. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [PubMed]
- Liais, E.; Croville, G.; Mariette, J.; Delverdier, M.; Lucas, M.N.; Klopp, C.; Lluch, J.; Donnadieu, C.; Guy, J.S.; Corrand, L.; et al. Novel avian coronavirus and fulminating disease in Guinea Fowl, France. Emerg. Infect. Dis. 2014, 20, 105–108. [Google Scholar] [CrossRef]
- Angly, F.E.; Willner, D.; Prieto-Davo, A.; Edwards, R.A.; Schmieder, R.; Vega-Thurber, R.; Antonopoulos, D.A.; Barott, K.; Cottrell, M.T.; Desnues, C.; et al. The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Ren, L.; Zhang, Y.; Li, J.; Xiao, Y.; Zhang, J.; Wang, Y.; Chen, L.; Paranhos-Baccalà, G.; Wang, J. Genetic drift of human coronavirus OC43 spike gene during adaptive evolution. Sci. Rep. 2015, 5, 11451. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Cotten, M.; Watson, S.J.; Zumla, A.I.; Makhdoom, H.Q.; Palser, A.L.; Ong, S.H.; Al Rabeeah, A.A.; Alhakeem, R.F.; Assiri, A.; Al-Tawfiq, J.A.; et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. MBio 2014, 5, e01062-13. [Google Scholar] [CrossRef]
- Storz, J.; Purdy, C.W.; Lin, X.; Burrell, M.; Truax, R.E.; Briggs, R.E.; Frank, G.H.; Loan, R.W. Isolation of respiratory bovine coronavirus, other cytocidal viruses, and Pasteurella spp from cattle involved in two natural outbreaks of shipping fever. J. Am. Vet. Med. Assoc. 2000, 216, 1599–1604. [Google Scholar] [CrossRef]
- Cho, K.O.; Hoet, A.E.; Loerch, S.C.; Wittum, T.E.; Saif, L.J. Evaluation of concurrent shedding of bovine coronavirus via the respiratory tract and enteric route in feedlot cattle. Am. J. Vet. Res. 2001, 62, 1436–1441. [Google Scholar] [CrossRef]
- Gulliksen, S.M.; Jor, E.; Lie, K.I.; Løken, T.; Åkerstedt, J.; Østerås, O. Respiratory infections in Norwegian dairy calves. J. Dairy Sci. 2009, 92, 5139–5146. [Google Scholar] [CrossRef]
- Pardon, B.; De Bleecker, K.; Dewulf, J.; Callens, J.; Boyen, F.; Catry, B.; Deprez, P. Prevalence of respiratory pathogens in diseased, non-vaccinated, routinely medicated veal calves. Vet. Rec. 2011, 169, 278. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.; Mooney, J.; Connaghan, E.; Furphy, C.; Graham, D.A. Patterns of detection of respiratory viruses in nasal swabs from calves in Ireland: A retrospective study. Vet. Rec. 2014, 175, 351. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Pelletier, C.; Herman, N.; Ducatez, M.; Cassard, H.; Salem, E. Vers une identification de nouveaux virus respiratoires bovins. NEVA 2015, 8, 80–86. [Google Scholar]
- Hasoksuz, M.; Lathrop, S.L.; Gadfield, K.L.; Saif, L.J. Isolation of bovine respiratory coronaviruses from feedlot cattle and comparison of their biological and antigenic properties with bovine enteric coronaviruses. Am. J. Vet. Res. 1999, 60, 1227–1233. [Google Scholar] [PubMed]
- Behura, S.K.; Tizioto, P.C.; Kim, J.; Grupioni, N.V.; Seabury, C.M.; Schnabel, R.D.; Gershwin, L.J.; Van Eenennaam, A.L.; Toaff-Rosenstein, R.; Neibergs, H.L.; et al. Tissue Tropism in Host Transcriptional Response to Members of the Bovine Respiratory Disease Complex. Sci. Rep. 2017, 7, 17938. [Google Scholar] [CrossRef]
- Valentova, V.; Antonis, A.F.G.; Kovarcik, K. Restriction enzyme analysis of RT-PCR amplicons as a rapid method for detection of genetic diversity among bovine respiratory syncytial virus isolates. Vet. Microbiol. 2005, 108, 1–12. [Google Scholar] [CrossRef]
- Sarmiento-Silva, R.E.; Nakamura-Lopez, Y.; Vaughan, G. Epidemiology, molecular epidemiology and evolution of bovine respiratory syncytial virus. Viruses 2012, 4, 3452–3467. [Google Scholar] [CrossRef]
- Valarcher, J.F.; Schelcher, F.; Bourhy, H. Evolution of bovine respiratory syncytial virus. J. Virol. 2000, 74, 10714–10728. [Google Scholar] [CrossRef]
- Vilcek, S.; Durkovic, B.; Kolesarova, M.; Paton, D.J. Genetic diversity of BVDV: Consequences for classification and molecular epidemiology. In Proceedings of the Preventive Veterinary Medicine; Elsevier: Amsterdam, The Netherlands, 2005; Volume 72, pp. 31–35. [Google Scholar]
- FAO statistics. Available online: http://www.fao.org/faostat/en/#data/TM (accessed on 11 April 2019).
- Collin, E.a.; Sheng, Z.; Lang, Y.; Ma, W.; Hause, B.M.; Li, F. Cocirculation of Two Distinct Genetic and Antigenic Lineages of Proposed Influenza D Virus in Cattle. J. Virol. 2015, 89, 1036–1042. [Google Scholar] [CrossRef]
- Ducatez, M.F.; Pelletier, C.; Meyer, G. Influenza D Virus in Cattle, France, 2011–2014. EID J. 2015, 21, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Flynn, O.; Gallagher, C.; Mooney, J.; Irvine, C.; Ducatez, M.; Hause, B.; McGrath, G.; Ryan, E. Influenza D virus in cattle, Ireland. Emerg. Infect. Dis. 2018, 24, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Foni, E.; Chiapponi, C.; Baioni, L.; Zanni, I.; Merenda, M.; Rosignoli, C.; Kyriakis, C.S.; Luini, M.V.; Mandola, M.L.; Nigrelli, A.D.; et al. Influenza D in Italy: Towards a better understanding of an emerging viral infection in swine. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Callow, K.A.; Parry, H.F.; Sergeant, M.; Tyrrell, D.A. The time course of the immune response to experimental coronavirus infection of man. Epidemiol. Infect. 1990, 105, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Bidokhti, M.R.M.; Tråvén, M.; Krishna, N.K.; Munir, M.; Belák, S.; Alenius, S.; Cortey, M. Evolutionary dynamics of bovine coronaviruses: Natural selection pattern of the spike gene implies adaptive evolution of the strains. J. Gen. Virol. 2013, 94, 2036–2049. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Lemey, P.; Maes, P.; Reeth, K.V.; Nauwynck, H.; Pensaert, M.; Ranst, M.V. Evolutionary History of the Closely Related Group 2 Coronaviruses: Porcine Hemagglutinating Encephalomyelitis Virus, Bovine Coronavirus, and Human Coronavirus OC43. J. Virol. 2006, 80, 7270–7274. [Google Scholar] [CrossRef]
- Sánchez, C.M.; Gebauer, F.; Suñé, C.; Mendez, A.; Dopazo, J.; Enjuanes, L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 1992, 190, 92–105. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Lee, P.; Tsang, A.K.L.; Yip, C.C.Y.; Tse, H.; Lee, R.A.; So, L.-Y.; Lau, Y.-L.; Chan, K.-H.; Woo, P.C.Y.; et al. Molecular Epidemiology of Human Coronavirus OC43 Reveals Evolution of Different Genotypes over Time and Recent Emergence of a Novel Genotype due to Natural Recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef]
- Salem, E. Bronchopneumonies Infectieuses des Jeunes Bovins: De la Complexité du Microbiome aux Particularités évolutives et Cliniques de virus Respiratoires Encore Méconnus. Ph.D. Thesis, INP Toulouse, Toulouse, France, 24 October 2018. [Google Scholar]



{kind=link}
{kind=link}
| Sample | Ct Value 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Id | Type 1 | P. multocida | M. haemolytica | M. bovis | H. somni | BCoV | RSV | PI-3 |
| ICSA-4 | BAL | 26.75 | 30.17 | 22.74 | ||||
| ICSA-16 | BAL | 39.01 | 24.19 | |||||
| ICSA-17 | BAL | 27.04 | 38.9 | |||||
| ICSA 21 | BAL | 29.32 | 27.31 | 29.27 | 38.61 | |||
| ICSA-4 | NS | 32.1 | 27.9 | 29.4 | 32.61 | |||
| ICSA-16 | NS | 18.67 | ||||||
| ICSA-17 | NS | 28.9 | ||||||
| ICSA 21 | NS | 24.4 | 26.7 | 34.93 | ||||
| Data | Gene | Demographic Model a | Mean nt Sub Rate (95% HPD) × 10−3 b | Mean tMRCA (95% HPD) in Years before 2015 |
|---|---|---|---|---|
| Global | S partition 1 | Constant | 0.8 (0.5–1) | 63.5 (53.9–75.9) |
| S partition 2 | Constant | 0.1 (0.7–1) | 60.94 (51.7–70.4) | |
| N | Constant | 0.2 (0.1–0.4) | 77.9 (46.2–123.2) | |
| Europe | S partition 1 | Constant | 0.7 (0.5–0.9) | 41.6 (37.5–53.5) |
| Bayesian skyline | 0.7 (0.5–0.9) | 41.9 (36.4–52.2) | ||
| S partition 2 | Constant | 0.8 (0.6–0.9) | 39. 7 (35.3–54.4) | |
| Bayesian skyline | 0.8 (0.6–1.0) | 37.5 (35.0–47.9) | ||
| N | Bayesian skyline | 0.5 (0.3–0.7) | 35.8 (35.0–45.4) | |
| BSP | 0.5 (0.3–0.7) | 35.6 (35.0–46.0 |
| Codon | SLAC p-Value | FEL p-Value | MEME p-Value |
|---|---|---|---|
| 35 | 0.0600 | 0.0700 | 0.0130 |
| 113 | 0.0340 | 0.0220 | 0.0700 |
| 115 | 0.0670 | 0.0210 | #N/A |
| 155 | #N/A | #N/A | 0.0500 |
| 179 | #N/A | #N/A | 0.0680 |
| 257 | #N/A | 0.0820 | #N/A |
| 304 | #N/A | #N/A | 0.0160 |
| 447 | 0.0260 | 0.0260 | 0.0660 |
| 484 | #N/A | #N/A | 0.0980 |
| 499 | 0.0230 | 0.0090 | #N/A |
| 501 | 0.0010 | 0.0040 | 0.0060 |
| 509 | #N/A | 0.0410 | #N/A |
| 525 | 0.0220 | 0.0280 | 0.0910 |
| 543 | #N/A | #N/A | 0.0760 |
| 689 | #N/A | #N/A | 0.0380 |
| 716 | 0.0590 | 0.0490 | #N/A |
| 744 | #N/A | #N/A | 0.0060 |
| 767 | #N/A | #N/A | 0.0000 |
| 769 | #N/A | #N/A | 0.0940 |
| 892 | #N/A | #N/A | 0.0710 |
| 893 | #N/A | 0.0410 | 0.0010 |
| 1085 | #N/A | #N/A | 0.0030 |
| 1188 | #N/A | #N/A | 0.0040 |
| 1206 | #N/A | 0.0450 | #N/A |
| 1237 | #N/A | #N/A | 0.0380 |
| 1239 | #N/A | 0.0110 | 0.0960 |
| 1269 | #N/A | #N/A | 0.0000 |
| 1362 | #N/A | 0.0150 | #N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, E.; Dhanasekaran, V.; Cassard, H.; Hause, B.; Maman, S.; Meyer, G.; Ducatez, M.F. Global Transmission, Spatial Segregation, and Recombination Determine the Long-Term Evolution and Epidemiology of Bovine Coronaviruses. Viruses 2020, 12, 534. https://doi.org/10.3390/v12050534
Salem E, Dhanasekaran V, Cassard H, Hause B, Maman S, Meyer G, Ducatez MF. Global Transmission, Spatial Segregation, and Recombination Determine the Long-Term Evolution and Epidemiology of Bovine Coronaviruses. Viruses. 2020; 12(5):534. https://doi.org/10.3390/v12050534
Chicago/Turabian StyleSalem, Elias, Vijaykrishna Dhanasekaran, Herve Cassard, Ben Hause, Sarah Maman, Gilles Meyer, and Mariette F. Ducatez. 2020. "Global Transmission, Spatial Segregation, and Recombination Determine the Long-Term Evolution and Epidemiology of Bovine Coronaviruses" Viruses 12, no. 5: 534. https://doi.org/10.3390/v12050534
APA StyleSalem, E., Dhanasekaran, V., Cassard, H., Hause, B., Maman, S., Meyer, G., & Ducatez, M. F. (2020). Global Transmission, Spatial Segregation, and Recombination Determine the Long-Term Evolution and Epidemiology of Bovine Coronaviruses. Viruses, 12(5), 534. https://doi.org/10.3390/v12050534