The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines


Abstract
{kind=link}
{kind=link}
{kind=link}
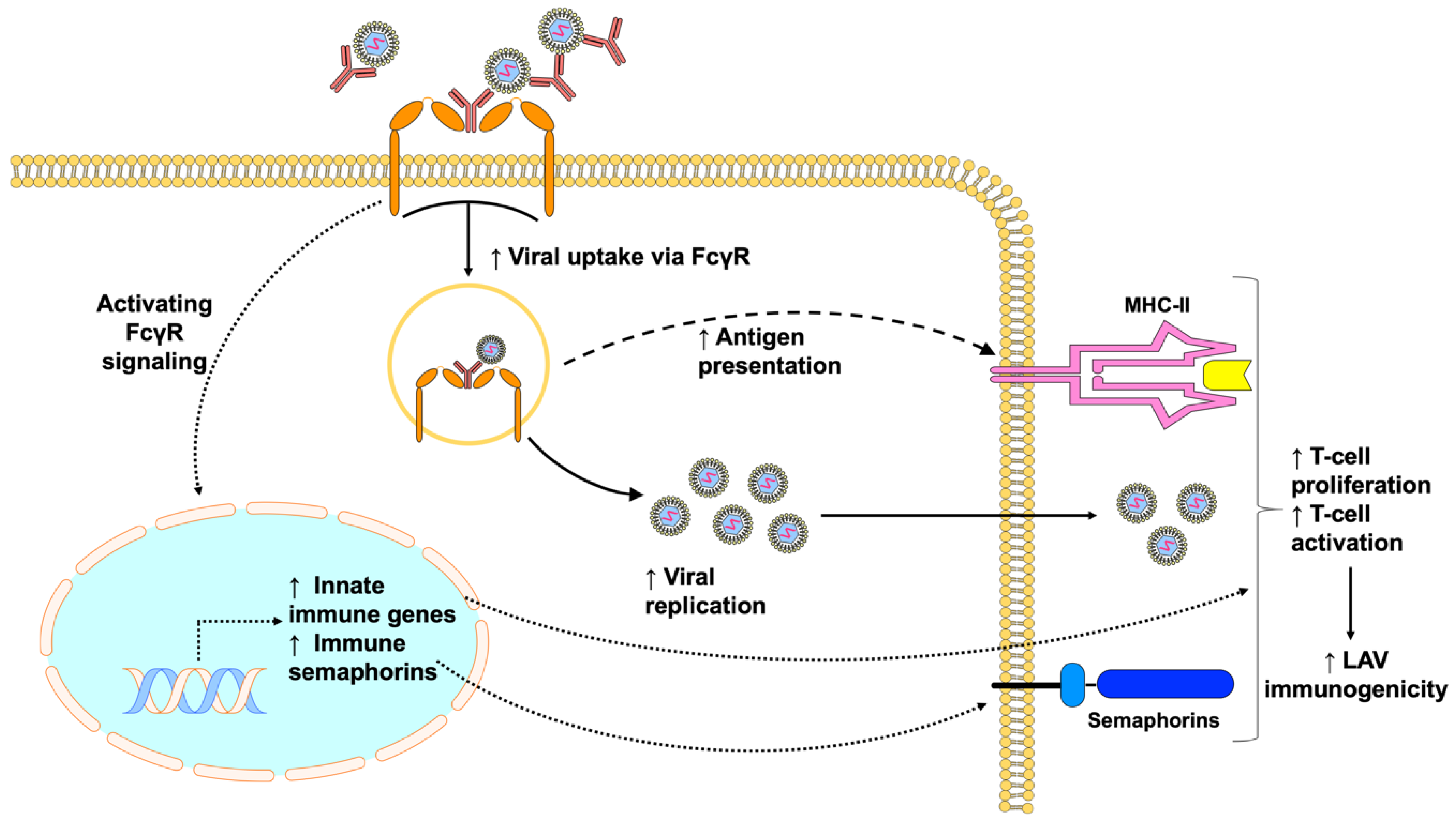{kind=link}
{kind=link}
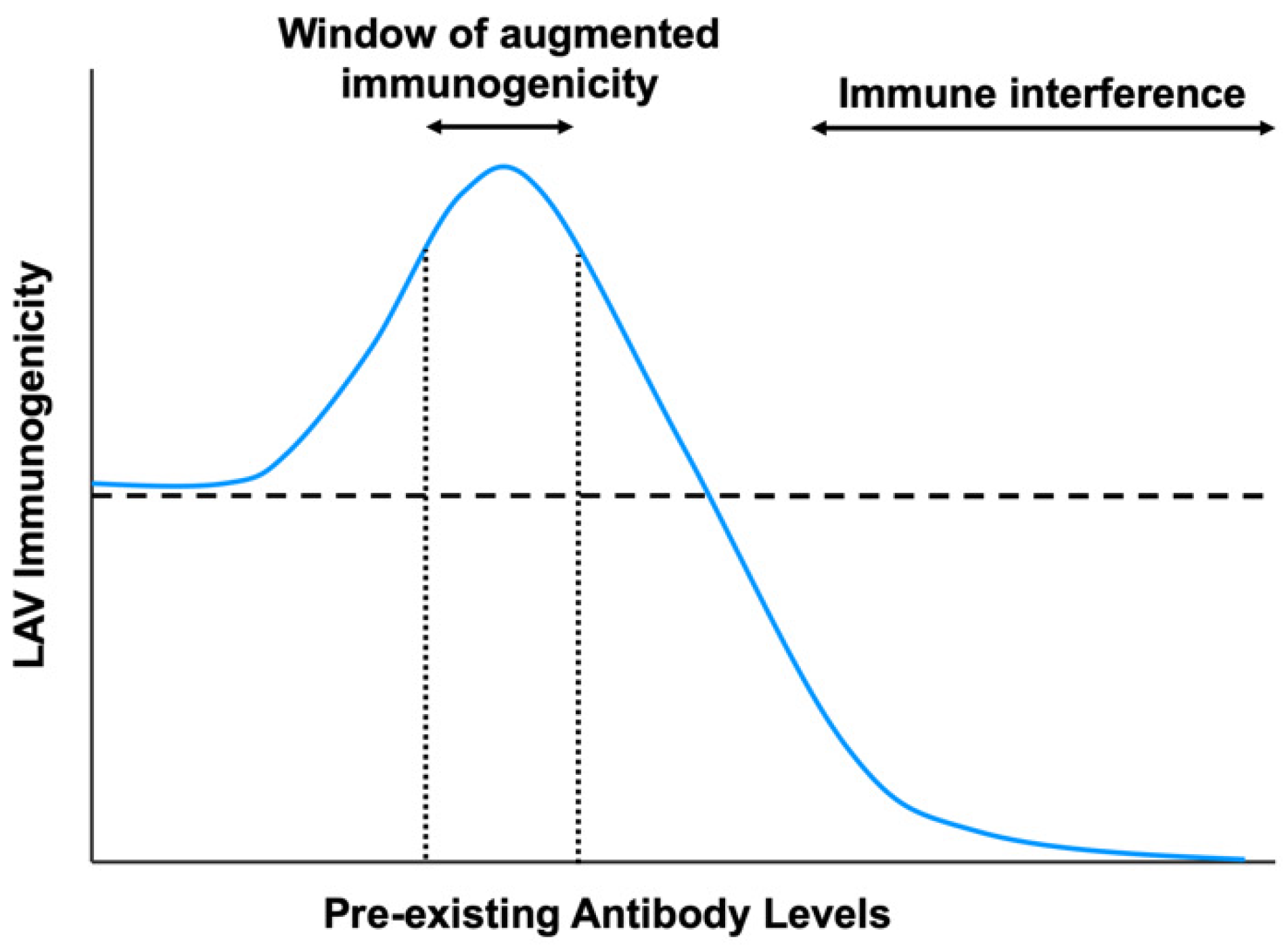
1. Introduction
2. Effects of Pre-Existing Immunity on Live Vaccines
2.1. Measles
2.2. Adenovirus and Adeno-Associated Virus
2.3. Influenza
2.4. Flaviviruses
2.5. Interaction of Antibody–Virus Complexes with Immune Cells
3. Mechanisms of Virus Inhibition Modulated by Pre-Existing Antibodies
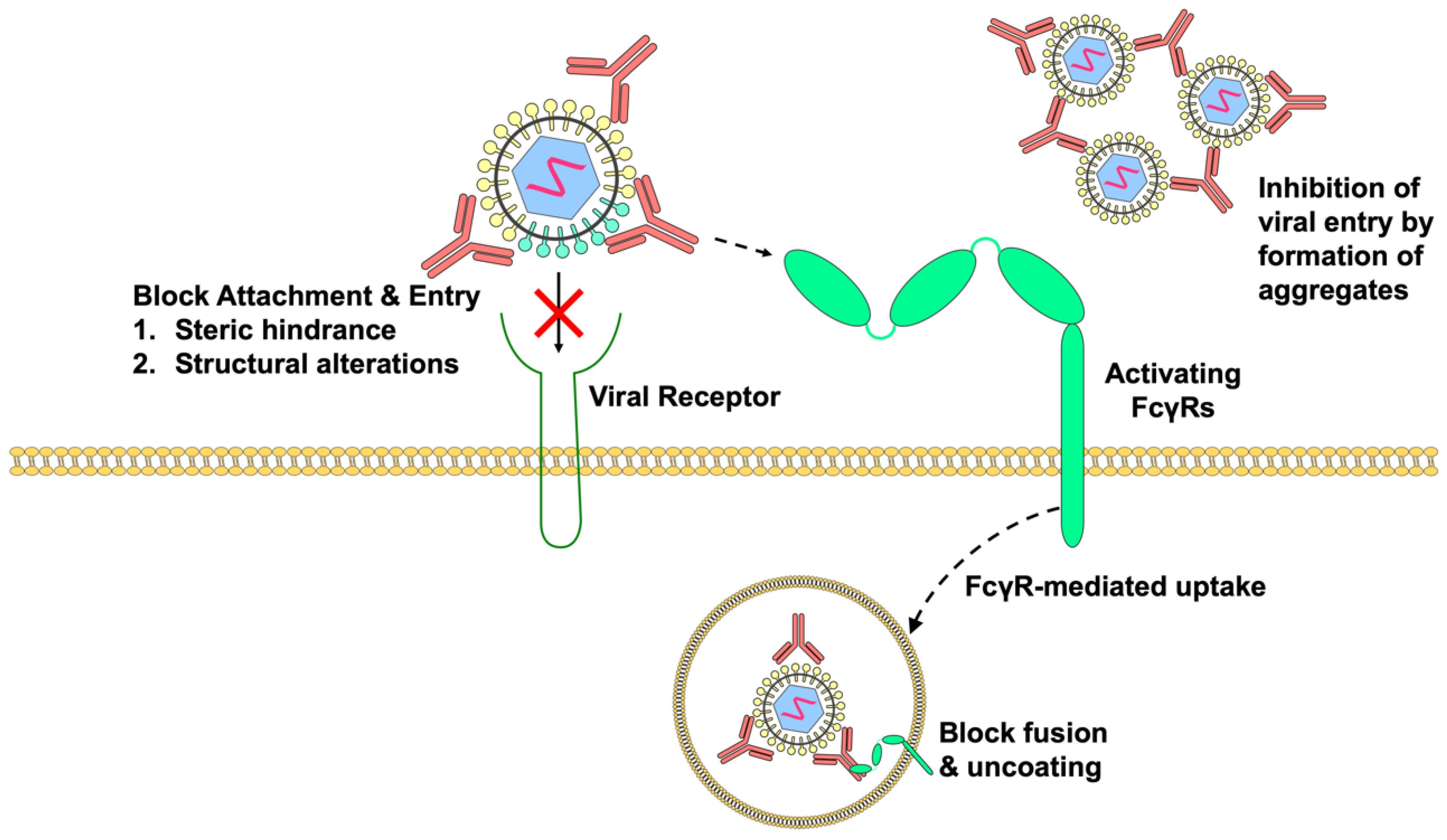
3.1. Neutralization of Live-Attenuated Vaccines
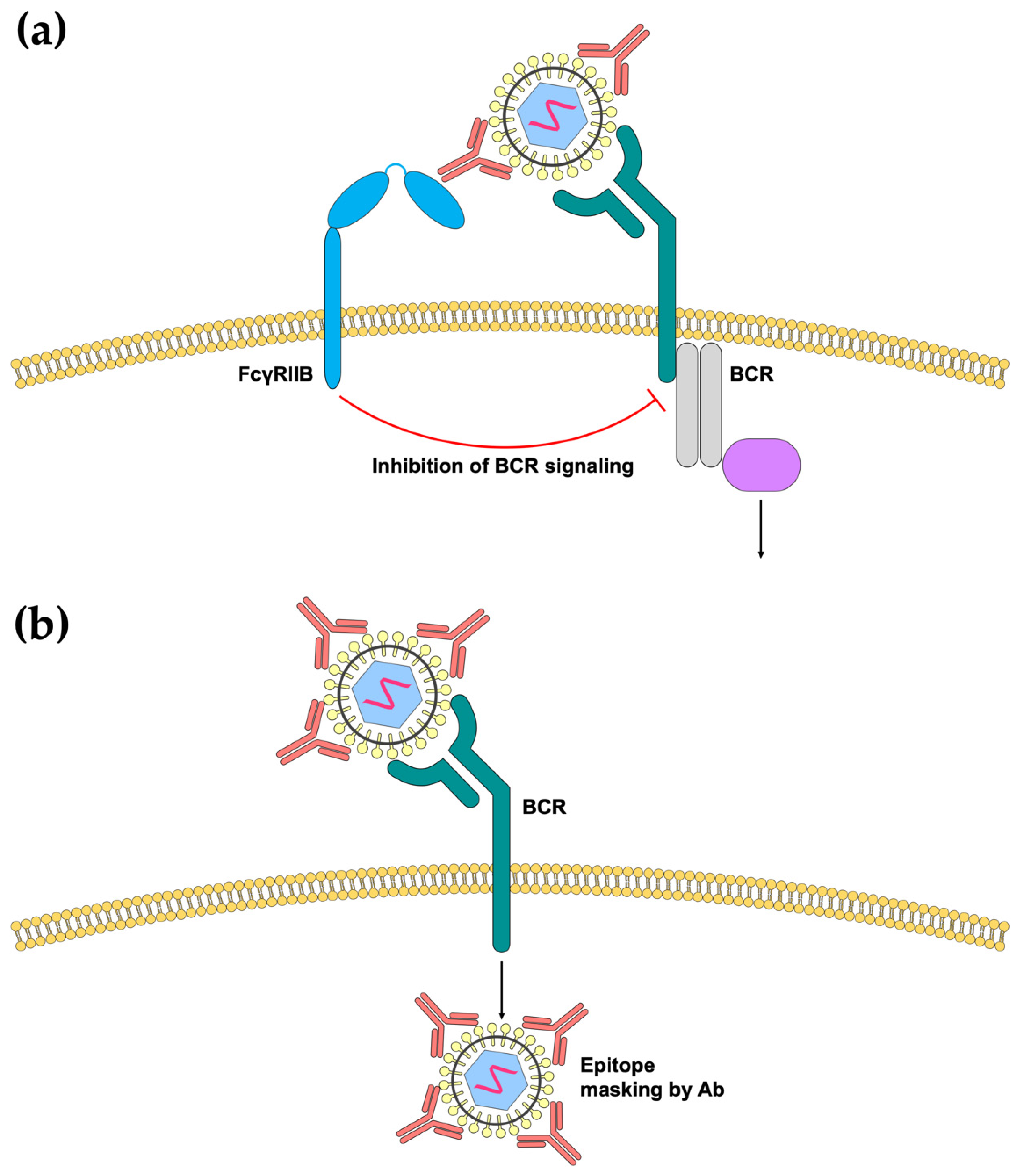
3.2. Inhibition of B-Cell Responses by Immune Complexes
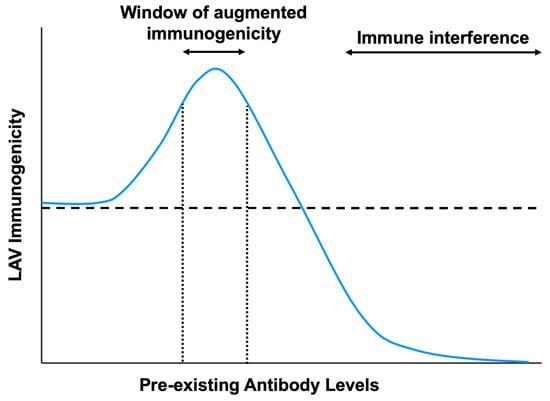
4. Mechanisms of Enhanced Vaccine Immunogenicity Modulated by Pre-Existing Antibodies
4.1. Antibody-Dependent Enhancement of LAV Infection and Immunogenicity
4.2. Intrinsic Host Responses That Promote Vaccine Immunogenicity
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Bristol, N. William H Stewart. The Lancet 2008, 372, 110. [Google Scholar] [CrossRef]
- Scalera, N.M.; Mossad, S.B. The First Pandemic of the 21st Century: Review of the 2009 Pandemic Variant Influenza A (H1N1) Virus. Postgrad. Med. 2009, 121, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Grobbelaar, A.A.; Weyer, J.; Moolla, N.; van Vuren, P.J.; Moises, F.; Paweska, J.T. Resurgence of Yellow Fever in Angola, 2015–2016. Emerg. Infect. Dis. 2016, 22, 1854. [Google Scholar] [CrossRef] [PubMed]
- Chowell, G.; Nishiura, H. Transmission dynamics and control of Ebola virus disease (EVD): A review. BMC Med. 2014, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Morens, D.M. Zika virus in the Americas—Yet another arbovirus threat. N. Engl. J. Med. 2016, 374, 601–604. [Google Scholar] [CrossRef]
- Lu, H.; Stratton, C.W.; Tang, Y.-W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef]
- Barrett, A.D.T.; Monath, T.P. Epidemiology and ecology of yellow fever virus. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2003; Volume 61, pp. 291–315. [Google Scholar]
- D’Argenio, D.A.; Wilson, C.B. A Decade of Vaccines: Integrating Immunology and Vaccinology for Rational Vaccine Design. Immunity 2010, 33, 437–440. [Google Scholar] [CrossRef]
- WHO. Declaration of global eradication of smallpox. Wkly. Epidemiol. Rec. 1980, 55, 148. [Google Scholar]
- WHO; World Bank. State of the World’s Vaccines and Immunization, Geneva; World Health Organization: Geneva, Switzerland, 2009; pp. 130–145. [Google Scholar]
- Stack, M.L.; Ozawa, S.; Bishai, D.M.; Mirelman, A.; Tam, Y.; Niessen, L.; Walker, D.G.; Levine, O.S. Estimated economic benefits during the ‘decade of vaccines’ include treatment savings, gains in labor productivity. Health Affairs 2011, 30, 1021–1028. [Google Scholar] [CrossRef]
- Benecke, O.; DeYoung, S.E. Anti-Vaccine Decision-Making and Measles Resurgence in the United States. Glob. Pediatr. Health 2019, 6, 2333794X19862949. [Google Scholar] [CrossRef]
- Siegrist, C.-A. Vaccine Immunology. In Vaccines; Saunders: Philadelphia, PA, USA, 2008; pp. 17–36. [Google Scholar]
- Jensen, P.E. Recent advances in antigen processing and presentation. Nat. Immunol. 2007, 8, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, H.B.; Arvin, A.M. Live attenuated vaccines: Influenza, rotavirus and varicella zoster virus. In Replicating Vaccines; Springer: Berlin, Germany, 2011; pp. 15–46. [Google Scholar]
- Humphreys, I.R.; Sebastian, S. Novel viral vectors in infectious diseases. Immunology 2018, 153, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Larocca, C.; Schlom, J. Viral vector-based therapeutic cancer vaccines. Cancer J. J. Princ. Pract. Oncol. 2011, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Guirakhoo, F.; Pugachev, K.; Zhang, Z.; Myers, G.; Levenbook, I.; Draper, K.; Lang, J.; Ocran, S.; Mitchell, F.; Parsons, M.; et al. Safety and Efficacy of Chimeric Yellow Fever-Dengue Virus Tetravalent Vaccine Formulations in Nonhuman Primates. J. Virol. 2004, 78, 4761. [Google Scholar] [CrossRef] [PubMed]
- Hadinegoro, S.R.; Arredondo-García, J.L.; Capeding, M.R.; Deseda, C.; Chotpitayasunondh, T.; Dietze, R.; Hj Muhammad Ismail, H.; Reynales, H.; Limkittikul, K.; Rivera-Medina, D.M. Efficacy and long-term safety of a dengue vaccine in regions of endemic disease. N. Engl. J. Med. 2015, 373, 1195–1206. [Google Scholar] [CrossRef]
- Gordon, S.B.; Rylance, J.; Luck, A.; Jambo, K.; Ferreira, D.M.; Manda-Taylor, L.; Bejon, P.; Ngwira, B.; Littler, K.; Seager, Z.J.W.o.r. A framework for Controlled Human Infection Model (CHIM) studies in Malawi: Report of a Wellcome Trust workshop on CHIM in Low Income Countries held in Blantyre, Malawi [version 1; peer review: 2 approved]. Wellcome Open Res. 2017, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Leuridan, E.; Hens, N.; Peeters, N.; de Witte, L.; Van der Meeren, O.; Van Damme, P. Effect of a prepregnancy pertussis booster dose on maternal antibody titers in young infants. Pediatr. Infect. Dis. J. 2011, 30, 608–610. [Google Scholar] [CrossRef]
- Leuridan, E.; Van Damme, P. Passive transmission and persistence of naturally acquired or vaccine-induced maternal antibodies against measles in newborns. Vaccine 2007, 25, 6296–6304. [Google Scholar] [CrossRef]
- Orenstein, W.A.; Perry, R.T.; Halsey, N.A. The clinical significance of measles: A review. J. Infect. Dis. 2004, 189, S4–S16. [Google Scholar] [CrossRef]
- Langmuir, A.D. Medical importance of measles. Am. J. Dis. Child. 1962, 103, 224–226. [Google Scholar] [CrossRef]
- Snyder, M.J.; McCrumb, F.R.; Bigbee, T.; Schluederberg, A.E.; Togo, Y. Observations on the seroepidemiology of measles. Am. J. Dis. Children 1962, 103, 250–251. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, M.; McQuaid, S.; Milner, D.; de Swart, R.L.; Duprex, W.P. Pathological consequences of systemic measles virus infection. J. Pathol. 2015, 235, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.P. The first measles vaccine. Pediatrics 2011, 128, 435–437. [Google Scholar] [CrossRef] [PubMed]
- CDC. Progress in global measles control and mortality reduction, 2000-2006. MMWR Morb. Mortal. Wkly. Rep. 2007, 56, 1237. [Google Scholar]
- Stein, C.E.; Birmingham, M.; Kurian, M.; Duclos, P.; Strebel, P. The global burden of measles in the year 2000—A model that uses country-specific indicators. J. Infect. Dis. 2003, 187, S8–S14. [Google Scholar] [CrossRef]
- Portnoy, A.; Jit, M.; Ferrari, M.; Hanson, M.; Brenzel, L.; Verguet, S. Estimates of case-fatality ratios of measles in low-income and middle-income countries: A systematic review and modelling analysis. Lancet Glob. Health 2019, 7, e472–e481. [Google Scholar] [CrossRef]
- Enders, J.F.; Peebles, T.C. Propagation in tissue cultures of cytopathogenic agents from patients with measles. Proc. Soc. Exp. Biol. Med. 1954, 86, 277–286. [Google Scholar] [CrossRef] [PubMed]
- WHO. Measles vaccines: WHO position paper. Wkly. Epidemiol. Rec. 2009, 84, 349–360. [Google Scholar]
- Itoh, M.; Okuno, Y.; Hotta, H. Comparative analysis of titers of antibody against measles virus in sera of vaccinated and naturally infected Japanese individuals of different age groups. J. Clin. Microbiol. 2002, 40, 1733–1738. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hinman, A.R.; Dine, M.S.; Hutchins, S.S.; Thomas, A.; Williams, I.; Bellini, W.J.; Redd, S.C. Persistence of vaccine-induced antibody to measles 26–33 years after vaccination. J. Infect. Dis. 2004, 189, S123–S130. [Google Scholar] [CrossRef]
- Knuchel, M.C.; Marty, R.R.; Morin, T.N.A.; Ilter, O.; Zuniga, A.; Naim, H.Y. Relevance of a pre-existing measles immunity prior immunization with a recombinant measles virus vector. Hum. Vaccines Immunother. 2013, 9, 599–606. [Google Scholar] [CrossRef]
- Kohler, P.F.; Farr, R.S. Elevation of cord over maternal IgG immunoglobulin: Evidence for an active placental IgG transport. Nature 1966, 210, 1070–1071. [Google Scholar] [CrossRef] [PubMed]
- Yeager, A.S.; Davis, J.H.; Ross, L.A.; Harvey, B. Measles immunization. Successes and failures. JAMA 1977, 237, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, P.; Ennis, F.A.; Saltzman, E.J.; Krugman, S. Persistence of maternal antibody in infants beyond 12 months: Mechanism of measles vaccine failure. J. Pediatr. 1977, 91, 715–718. [Google Scholar] [CrossRef]
- King, G.E.; Markowitz, L.E.; Patriarca, P.A.; Dales, L.G. Clinical efficacy of measles vaccine during the 1990 measles epidemic. Pediatr. Infect. Dis. J. 1991, 10, 883–888. [Google Scholar] [CrossRef]
- Orenstein, W.; Markowitz, L.; Preblud, S.; Hinman, A.; Tomasi, A.; Bart, K. Appropriate age for measles vaccination in the United States. Dev. Biol. Stand. 1986, 65, 13–21. [Google Scholar]
- Aaby, P.; Martins, C.L.; Garly, M.-L.; Andersen, A.; Fisker, A.B.; Claesson, M.H.; Ravn, H.; Rodrigues, A.; Whittle, H.C.; Benn, C.S. Measles vaccination in the presence or absence of maternal measles antibody: Impact on child survival. Clin. Infect. Dis. 2014, 59, 484–492. [Google Scholar] [CrossRef]
- Van Binnendijk, R.S.; Poelen, M.C.; van Amerongen, G.; de Vries, P.; Osterhaus, A.D. Protective immunity in macaques vaccinated with live attenuated, recombinant, and subunit measles vaccines in the presence of passively acquired antibodies. J. Infect. Dis. 1997, 175, 524–532. [Google Scholar] [CrossRef]
- Gans, H.A.; Ren, J.; Yasukawa, L.L.; Alderson, A.; Rinki, M.; DeHovitz, R.; Beeler, J.; Audet, S.; Maldonado, Y.; Arvin, A.M. Humoral and cell-mediated immune responses to an early 2-dose measles vaccination regimen in the United States. J. Infect. Dis. 2004, 190, 83–90. [Google Scholar] [CrossRef]
- Chirmule, N.; Propert, K.; Magosin, S.; Qian, Y.; Qian, R.; Wilson, J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Therapy 1999, 6, 1574–1583. [Google Scholar] [CrossRef]
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: Correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine 2010, 28, 950–957. [Google Scholar] [CrossRef]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; del Rio, C. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef]
- Yang, Z.-y.; Wyatt, L.S.; Kong, W.-p.; Moodie, Z.; Moss, B.; Nabel, G.J. Overcoming immunity to a viral vaccine by DNA priming before vector boosting. J. Virol. 2003, 77, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, D.R.; Chen, L.; Fu, T.-M.; Evans, R.K.; Caulfield, M.J.; Davies, M.-E.; Tang, A.; Chen, M.; Huang, L.; Harris, V. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J. Virol. 2003, 77, 6305–6313. [Google Scholar] [CrossRef] [PubMed]
- Smaill, F.; Jeyanathan, M.; Smieja, M.; Medina, M.F.; Thanthrige-Don, N.; Zganiacz, A.; Yin, C.; Heriazon, A.; Damjanovic, D.; Puri, L. A human type 5 adenovirus–based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci. Transl. Med. 2013, 5, 205ra134. [Google Scholar] [CrossRef]
- Gabitzsch, E.; Jones, F. New recombinant Ad5 vector overcomes Ad5 immunity allowing for multiple safe, homologous immunizations. J. Clin. Cell. Immunol. 2011, 4, 001. [Google Scholar] [CrossRef]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef]
- Heemskerk, B.; Veltrop-Duits, L.A.; van Vreeswijk, T.; Monique, M.; Heidt, S.; Toes, R.E.; van Tol, M.J.; Schilham, M.W. Extensive cross-reactivity of CD4+ adenovirus-specific T cells: Implications for immunotherapy and gene therapy. J. Virol. 2003, 77, 6562–6566. [Google Scholar] [CrossRef]
- Barouch, D.H.; Pau, M.G.; Custers, J.H.; Koudstaal, W.; Kostense, S.; Havenga, M.J.; Truitt, D.M.; Sumida, S.M.; Kishko, M.G.; Arthur, J.C. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J. Immunol. 2004, 172, 6290–6297. [Google Scholar] [CrossRef]
- Lemckert, A.A.; Sumida, S.M.; Holterman, L.; Vogels, R.; Truitt, D.M.; Lynch, D.M.; Nanda, A.; Ewald, B.A.; Gorgone, D.A.; Lifton, M.A. Immunogenicity of heterologous prime-boost regimens involving recombinant adenovirus serotype 11 (Ad11) and Ad35 vaccine vectors in the presence of anti-ad5 immunity. J. Virol. 2005, 79, 9694–9701. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- WHO. Global Influenza Strategy 2019–2030; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Grohskopf, L.A.; Alyanak, E.; Broder, K.R.; Walter, E.B.; Fry, A.M.; Jernigan, D.B. Prevention and Control of Seasonal Influenza with Vaccines: Recommendations of the Advisory Committee on Immunization Practices - United States, 2019-20 Influenza Season. MMWR Recomm. Rep. 2019, 68, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Houser, K.; Subbarao, K. Influenza vaccines: Challenges and solutions. Cell Host Microbe 2015, 17, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, C.S.; Wu, X.; Jones, T.; Mallory, R.M. The role of nasal IgA in children vaccinated with live attenuated influenza vaccine. Vaccine 2012, 30, 6794–6801. [Google Scholar] [CrossRef]
- Forrest, B.D.; Pride, M.W.; Dunning, A.J.; Capeding, M.R.Z.; Chotpitayasunondh, T.; Tam, J.S.; Rappaport, R.; Eldridge, J.H.; Gruber, W.C. Correlation of Cellular Immune Responses with Protection against Culture-Confirmed Influenza Virus in Young Children. Clin. Vaccine Immunol. 2008, 15, 1042–1053. [Google Scholar] [CrossRef]
- Ambrose, C.S.; Wu, X.; Knuf, M.; Wutzler, P. The efficacy of intranasal live attenuated influenza vaccine in children 2 through 17 years of age: A meta-analysis of 8 randomized controlled studies. Vaccine 2012, 30, 886–892. [Google Scholar] [CrossRef]
- Brickley, E.B.; Wright, P.F.; Khalenkov, A.; Neuzil, K.M.; Ortiz, J.R.; Rudenko, L.; Levine, M.Z.; Katz, J.M.; Brooks, W.A. The Effect of Preexisting Immunity on Virus Detection and Immune Responses in a Phase II, Randomized Trial of a Russian-Backbone, Live, Attenuated Influenza Vaccine in Bangladeshi Children. Clin. Infect. Dis. 2019, 69, 786–794. [Google Scholar] [CrossRef]
- Mohn, K.G.; Bredholt, G.; Brokstad, K.A.; Pathirana, R.D.; Aarstad, H.J.; Tondel, C.; Cox, R.J. Longevity of B-cell and T-cell responses after live attenuated influenza vaccination in children. J. Infect. Dis. 2015, 211, 1541–1549. [Google Scholar] [CrossRef]
- Mohn, K.G.I.; Zhou, F.; Brokstad, K.A.; Sridhar, S.; Cox, R.J. Boosting of Cross-Reactive and Protection-Associated T Cells in Children After Live Attenuated Influenza Vaccination. J. Infect. Dis. 2017, 215, 1527–1535. [Google Scholar] [CrossRef]
- Gruber, W.C.; Darden, P.M.; Still, J.G.; Lohr, J.; Reed, G.; Wright, P.F. Evaluation of bivalent live attenuated influenza A vaccines in children 2 months to 3 years of age: Safety, immunogenicity and dose-response. Vaccine 1997, 15, 1379–1384. [Google Scholar] [CrossRef]
- Saito, N.; Komori, K.; Suzuki, M.; Morimoto, K.; Kishikawa, T.; Yasaka, T.; Ariyoshi, K. Negative impact of prior influenza vaccination on current influenza vaccination among people infected and not infected in prior season: A test-negative case-control study in Japan. Vaccine 2017, 35, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; He, X.S.; Holmes, T.H.; Dekker, C.L.; Kemble, G.W.; Arvin, A.M.; Greenberg, H.B. Influence of prior influenza vaccination on antibody and B-cell responses. PLoS ONE 2008, 3, e2975. [Google Scholar] [CrossRef] [PubMed]
- Coelingh, K.L.; Luke, C.J.; Jin, H.; Talaat, K.R. Development of live attenuated influenza vaccines against pandemic influenza strains. Expert Rev. Vaccines 2014, 13, 855–871. [Google Scholar] [CrossRef] [PubMed]
- Coelingh, K.L.; Wu, X.W.; Mallory, R.M.; Ambrose, C.S. An integrated multi-study analysis of serum HAI antibody responses to Ann Arbor strain live attenuated influenza vaccine in children and adults. Trials Vaccinol. 2014, 3, 150–153. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Musso, D.; Ko, A.I.; Baud, D. Zika Virus Infection—After the Pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Calisher, C.H.; Karabatsos, N.; Dalrymple, J.M.; Shope, R.E.; Porterfield, J.S.; Westaway, E.G.; Brandt, W.E. Antigenic relationships between flaviviruses as determined by cross-neutralization tests with polyclonal antisera. J. General Virol. 1989, 70, 37–43. [Google Scholar] [CrossRef]
- Rathore, A.P.S.; St John, A.L. Cross-Reactive Immunity Among Flaviviruses. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Snow, G.E.; Haaland, B.; Ooi, E.E.; Gubler, D.J. Review article: Research on dengue during World War II revisited. Am. J. Trop. Med. Hyg. 2014, 91, 1203–1217. [Google Scholar] [CrossRef]
- Sabin, A.B. Research on dengue during World War II. Am. J. Trop. Med. Hyg. 1952, 1, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.G.; Kouri, G.; Valdes, L.; Bravo, J.; Alvarez, M.; Vazques, S.; Delgado, I.; Halstead, S.B. Epidemiologic Studies on Dengue in Santiago de Cuba, 1997. Am. J. Epidemiol. 2000, 152, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef]
- Salje, H.; Cummings, D.A.T.; Rodriguez-Barraquer, I.; Katzelnick, L.C.; Lessler, J.; Klungthong, C.; Thaisomboonsuk, B.; Nisalak, A.; Weg, A.; Ellison, D.; et al. Reconstruction of antibody dynamics and infection histories to evaluate dengue risk. Nature 2018, 557, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Kliks, S.C.; Nimmanitya, S.; Nisalak, A.; Burke, D.S. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am. J. Trop. Med. Hyg. 1988, 38, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.; Dayan, G.H.; Arredondo-García, J.L.; Rivera, D.M.; Cunha, R.; Deseda, C.; Reynales, H.; Costa, M.S.; Morales-Ramírez, J.O.; Carrasquilla, G. Efficacy of a tetravalent dengue vaccine in children in Latin America. N. Engl. J. Med. 2015, 372, 113–123. [Google Scholar] [CrossRef]
- Biswal, S.; Reynales, H.; Saez-Llorens, X.; Lopez, P.; Borja-Tabora, C.; Kosalaraksa, P.; Sirivichayakul, C.; Watanaveeradej, V.; Rivera, L.; Espinoza, F.; et al. Efficacy of a Tetravalent Dengue Vaccine in Healthy Children and Adolescents. N. Engl. J. Med. 2019, 381, 2009–2019. [Google Scholar] [CrossRef]
- Saron, W.A.A.; Rathore, A.P.S.; Ting, L.; Ooi, E.E.; Low, J.; Abraham, S.N.; St John, A.L. Flavivirus serocomplex cross-reactive immunity is protective by activating heterologous memory CD4 T cells. Science Adv. 2018, 4, eaar4297. [Google Scholar] [CrossRef]
- Bradt, V.; Malafa, S.; von Braun, A.; Jarmer, J.; Tsouchnikas, G.; Medits, I.; Wanke, K.; Karrer, U.; Stiasny, K.; Heinz, F.X. Pre-existing yellow fever immunity impairs and modulates the antibody response to tick-borne encephalitis vaccination. npj Vaccines 2019, 4, 38. [Google Scholar] [CrossRef]
- Chan, K.R.; Wang, X.; Saron, W.A.; Gan, E.S.; Tan, H.C.; Mok, D.Z.; Zhang, S.L.; Lee, Y.H.; Liang, C.; Wijaya, L.; et al. Cross-reactive antibodies enhance live attenuated virus infection for increased immunogenicity. Nat. Microbiol. 2016, 1, 16164. [Google Scholar] [CrossRef]
- Ravetch, J.V.; Bolland, S. IgG Fc Receptors. Ann. Rev. Immunol. 2001, 19, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Bolen, J.B.; Fleit, H.B. Physical and functional association of Src-related protein tyrosine kinases with Fc gamma RII in monocytic THP-1 cells. J. Biol. Chem. 1994, 269, 8878–8884. [Google Scholar] [PubMed]
- Crowley, M.T.; Costello, P.S.; Fitzer-Attas, C.J.; Turner, M.; Meng, F.; Lowell, C.; Tybulewicz, V.L.; DeFranco, A.L. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 1997, 186, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Cutler, A.J.; Brownlie, R.J.; Fairfax, K.; Lawlor, K.E.; Severinson, E.; Walker, E.U.; Manz, R.A.; Tarlinton, D.M.; Smith, K.G. FcγRIIb controls bone marrow plasma cell persistence and apoptosis. Nat. Immunol. 2007, 8, 419–429. [Google Scholar] [CrossRef]
- Mackay, M.; Stanevsky, A.; Wang, T.; Aranow, C.; Li, M.; Koenig, S.; Ravetch, J.V.; Diamond, B. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J. Exp. Med. 2006, 203, 2157–2164. [Google Scholar] [CrossRef]
- Smith, K.G.; Clatworthy, M.R. FcγRIIB in autoimmunity and infection: Evolutionary and therapeutic implications. Nat. Rev. Immunol. 2010, 10, 328–343. [Google Scholar] [CrossRef]
- Carter, N.A.; Harnett, M.M. Dissection of the signalling mechanisms underlying FcγRIIB-mediated apoptosis of mature B-cells. Biochem. Soc. Trans. 2004, 32, 973–975. [Google Scholar] [CrossRef]
- Tzeng, S.-J.; Bolland, S.; Inabe, K.; Kurosaki, T.; Pierce, S.K. The B cell inhibitory Fc receptor triggers apoptosis by a novel c-Abl family kinase-dependent pathway. J. Biol. Chem. 2005, 280, 35247–35254. [Google Scholar] [CrossRef]
- Tackenberg, B.; Jelčić, I.; Baerenwaldt, A.; Oertel, W.H.; Sommer, N.; Nimmerjahn, F.; Lünemann, J.D. Impaired inhibitory Fcγ receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc. Natl. Acad. Sci. 2009, 106, 4788–4792. [Google Scholar] [CrossRef]
- Tzeng, S.-J.; Li, W.-Y.; Wang, H.-Y. FcγRIIB mediates antigen-independent inhibition on human B lymphocytes through Btk and p38 MAPK. J. Biomed. Sci. 2015, 22, 87. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. Molecular mechanisms of antibody-mediated neutralisation of flavivirus infection. Expert Rev. Mol. Med. 2008, 10, e12. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Xu, Q.; Nelson, S.; Oliphant, T.; Nybakken, G.E.; Fremont, D.H.; Diamond, M.S. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 2007, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Fremont, D.H.; Kuhn, R.J.; Diamond, M.S. Structural Insights into the Mechanisms of Antibody-Mediated Neutralization of Flavivirus Infection: Implications for Vaccine Development. Cell Host Microbe 2008, 4, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; Pierson, T.C. Antibody-mediated neutralization of flaviviruses: A reductionist view. Virology 2011, 411, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lee, L.Y.; Roivainen, M.; Filman, D.J.; Hogle, J.M.; Belnap, D.M. Structure of the Fab-labeled “breathing” state of native poliovirus. J. Virol. 2012, 86, 5959–5962. [Google Scholar] [CrossRef]
- Chan, K.R.; Ong, E.Z.; Mok, D.Z.; Ooi, E.E. Fc receptors and their influence on efficacy of therapeutic antibodies for treatment of viral diseases. Expert Rev. Anti-Infect. Ther. 2015, 13, 1351–1360. [Google Scholar] [CrossRef]
- Chan, K.R.; Zhang, S.L.-X.; Tan, H.C.; Chan, Y.K.; Chow, A.; Lim, A.P.C.; Vasudevan, S.G.; Hanson, B.J.; Ooi, E.E. Ligation of Fc gamma receptor IIB inhibits antibody-dependent enhancement of dengue virus infection. Proc. Natl. Acad. Sci. 2011, 108, 12479–12484. [Google Scholar] [CrossRef]
- Halstead, S.B. Pathogenic Exploitation of Fc Activity. Antibody Fc 2014. [Google Scholar] [CrossRef]
- Karlsson, M.C.; Getahun, A.; Heyman, B. FcgammaRIIB in IgG-mediated suppression of antibody responses: Different impact in vivo and in vitro. J. Immunol. 2001, 167, 5558–5564. [Google Scholar] [CrossRef]
- Kim, D.; Huey, D.; Oglesbee, M.; Niewiesk, S. Insights into the regulatory mechanism controlling the inhibition of vaccine-induced seroconversion by maternal antibodies. Blood 2011, 117, 6143–6151. [Google Scholar] [CrossRef]
- Siegrist, C.A.; Plotnicky-Gilquin, H.; Cordova, M.; Berney, M.; Bonnefoy, J.Y.; Nguyen, T.N.; Lambert, P.H.; Power, U.F. Protective efficacy against respiratory syncytial virus following murine neonatal immunization with BBG2Na vaccine: Influence of adjuvants and maternal antibodies. J. Infect. Dis. 1999, 179, 1326–1333. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Getahun, A.; Heyman, B. Studies on the Mechanism by Which Antigen-Specific IgG Suppresses Primary Antibody Responses: Evidence for Epitope Masking and Decreased Localization of Antigen in the Spleen. Scand. J. Immunol. 2009, 70, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Heyman, B.; Wigzell, H. Immunoregulation by monoclonal sheep erythrocyte-specific IgG antibodies: Suppression is correlated to level of antigen binding and not to isotype. J. Immunol. 1984, 132, 1136–1143. [Google Scholar] [PubMed]
- Halstead, S.B.; Mahalingam, S.; Marovich, M.A.; Ubol, S.; Mosser, D.M. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: Disease regulation by immune complexes. Lancet Infect. Dis. 2010, 10, 712–722. [Google Scholar] [CrossRef]
- Chawla, T.; Chan, K.R.; Zhang, S.L.; Tan, H.C.; Lim, A.P.C.; Hanson, B.J.; Ooi, E.E. Dengue Virus Neutralization in Cells Expressing Fc Gamma Receptors. PLoS ONE 2013, 8, e65231. [Google Scholar] [CrossRef]
- Rodrigo, W.W.S.I.; Jin, X.; Blackley, S.D.; Rose, R.C.; Schlesinger, J.J. Differential Enhancement of Dengue Virus Immune Complex Infectivity Mediated by Signaling-Competent and Signaling-Incompetent Human FcγRIA (CD64) or FcγRIIA (CD32). J. Virol. 2006, 80, 10128–10138. [Google Scholar] [CrossRef]
- Dai, X.; Jayapal, M.; Tay, H.K.; Reghunathan, R.; Lin, G.; Too, C.T.; Lim, Y.T.; Chan, S.H.; Kemeny, D.M.; Floto, R.A.; et al. Differential signal transduction, membrane trafficking, and immune effector functions mediated by FcγRI versus FcγRIIa. Blood 2009, 114, 318–327. [Google Scholar] [CrossRef]
- Flipse, J.; Diosa-Toro, M.A.; Hoornweg, T.E.; van de Pol, D.P.I.; Urcuqui-Inchima, S.; Smit, J.M. Antibody-Dependent Enhancement of Dengue Virus Infection in Primary Human Macrophages; Balancing Higher Fusion against Antiviral Responses. Sci. Rep. 2016, 6, 29201. [Google Scholar] [CrossRef]
- Kumanogoh, A.; Marukawa, S.; Suzuki, K.; Takegahara, N.; Watanabe, C.; Ch’ng, E.; Ishida, I.; Fujimura, H.; Sakoda, S.; Yoshida, K.; et al. Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature 2002, 419, 629–633. [Google Scholar] [CrossRef]
- Kumanogoh, A.; Shikina, T.; Suzuki, K.; Uematsu, S.; Yukawa, K.; Kashiwamura, S.-I.; Tsutsui, H.; Yamamoto, M.; Takamatsu, H.; Ko-Mitamura, E.P.; et al. Nonredundant Roles of Sema4A in the Immune System: Defective T Cell Priming and Th1/Th2 Regulation in Sema4A-Deficient Mice. Immunity 2005, 22, 305–316. [Google Scholar] [CrossRef]
- Chan, K.R.; Gan, E.S.; Chan, C.Y.Y.; Liang, C.; Low, J.Z.H.; Zhang, S.L.-X.; Ong, E.Z.; Bhatta, A.; Wijaya, L.; Lee, Y.H. Metabolic perturbations and cellular stress underpin susceptibility to symptomatic live-attenuated yellow fever infection. Nat. Med. 2019, 25, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Kotliarov, Y.; Sparks, R.; Martins, A.J.; Mulè, M.P.; Lu, Y.; Goswami, M.; Kardava, L.; Banchereau, R.; Pascual, V.; Biancotto, A.J.N.M. Broad immune activation underlies shared set point signatures for vaccine responsiveness in healthy individuals and disease activity in patients with lupus. Nat. Med. 2020, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mok, D.Z.L.; Chan, K.R. The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines. Viruses 2020, 12, 520. https://doi.org/10.3390/v12050520
Mok DZL, Chan KR. The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines. Viruses. 2020; 12(5):520. https://doi.org/10.3390/v12050520
Chicago/Turabian StyleMok, Darren Z. L., and Kuan Rong Chan. 2020. "The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines" Viruses 12, no. 5: 520. https://doi.org/10.3390/v12050520
APA StyleMok, D. Z. L., & Chan, K. R. (2020). The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines. Viruses, 12(5), 520. https://doi.org/10.3390/v12050520