Genomic Analyses of Human Sapoviruses Detected over a 40-Year Period Reveal Disparate Patterns of Evolution among Genotypes and Genome Regions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fecal Samples Positive for Human Sapovirus
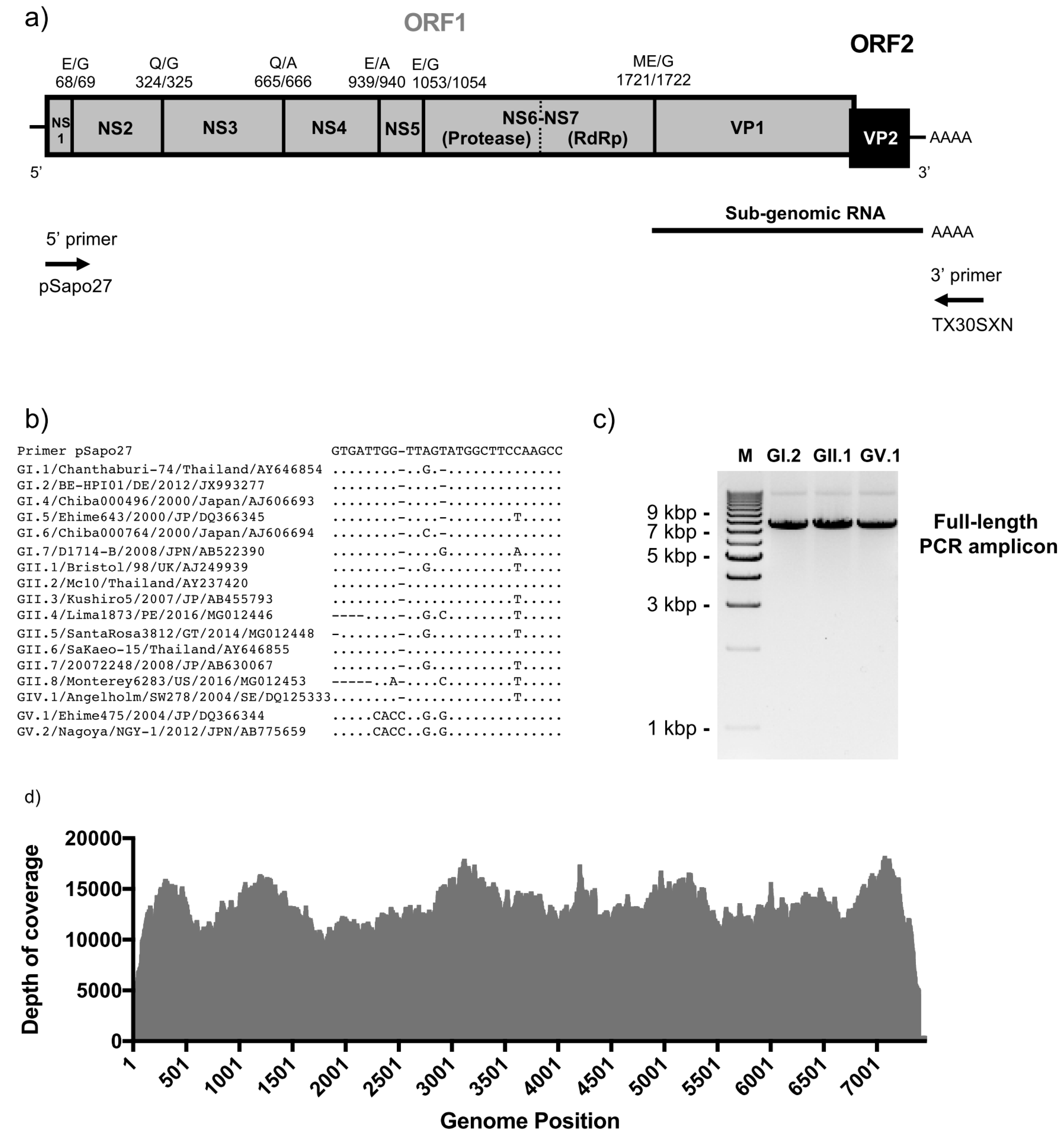
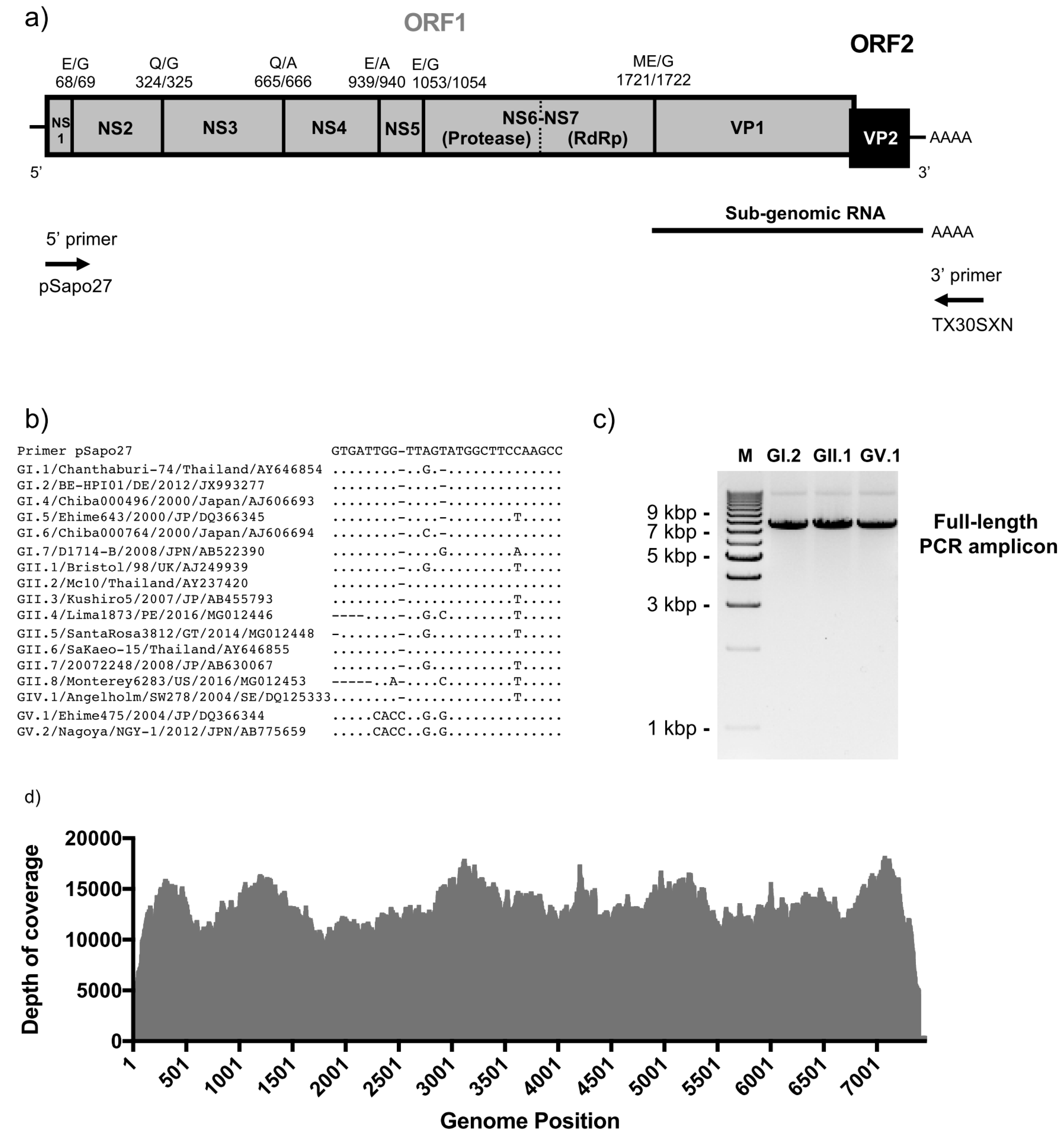
2.2. Full-Length Viral Genome PCR and Sequencing
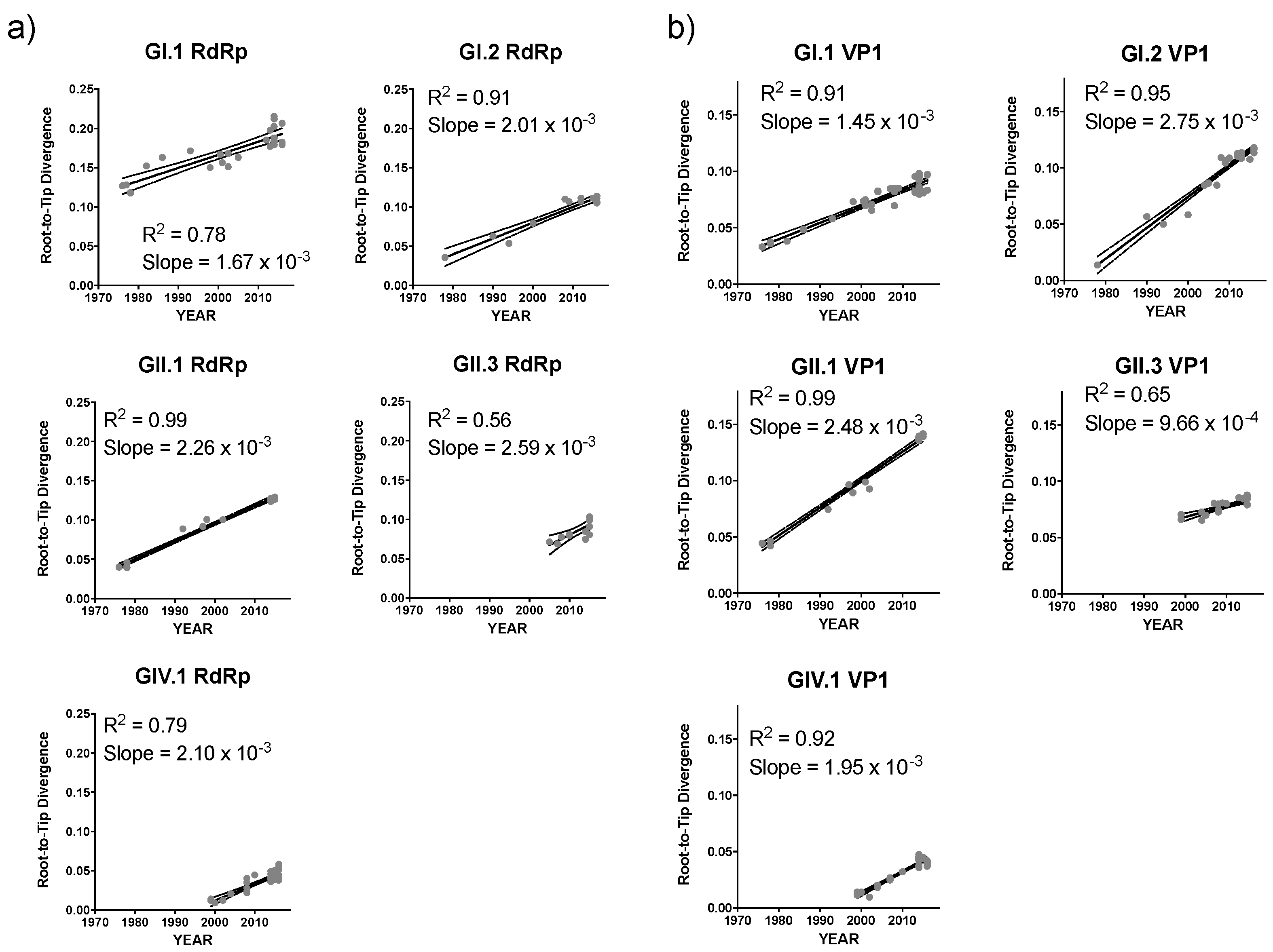
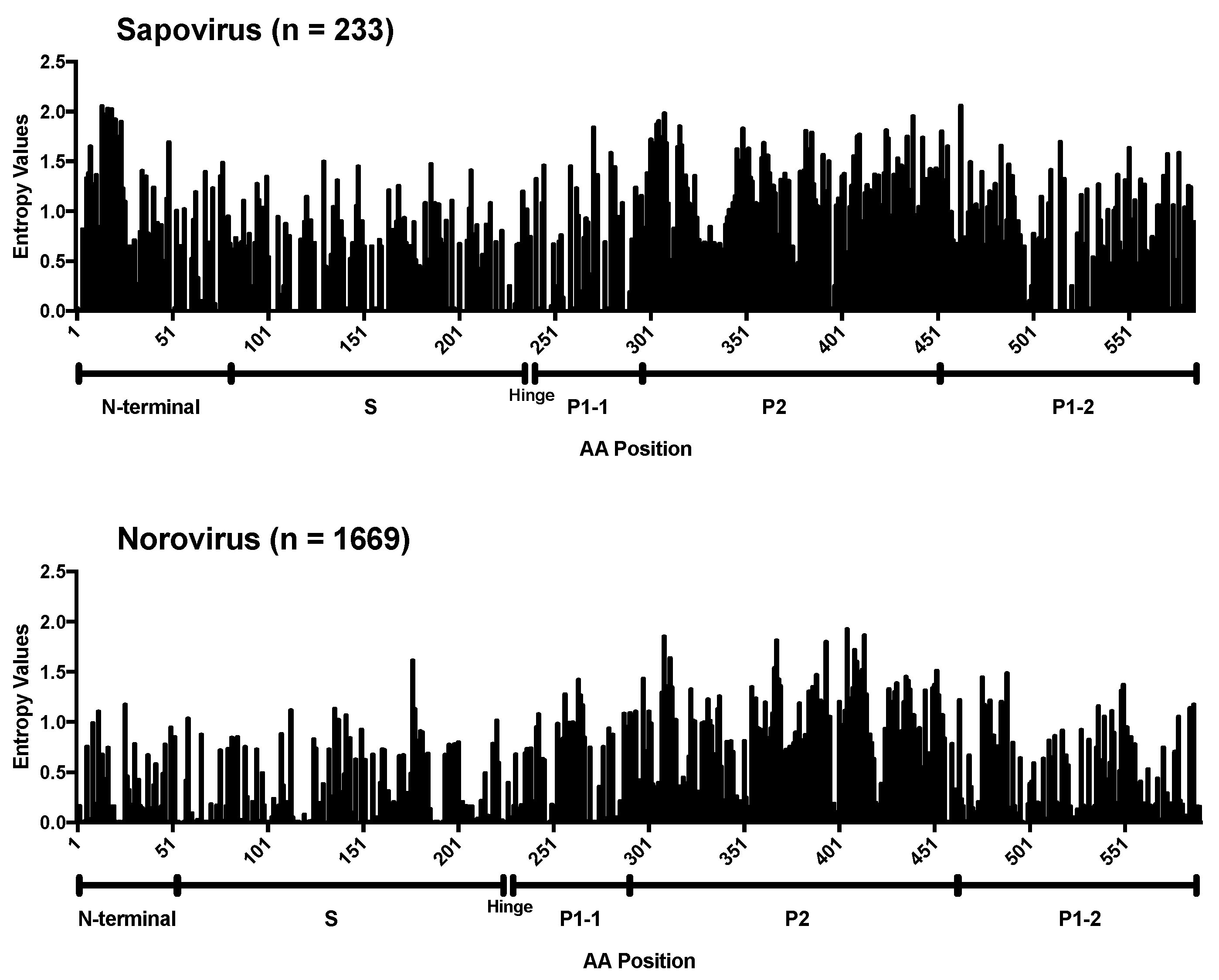
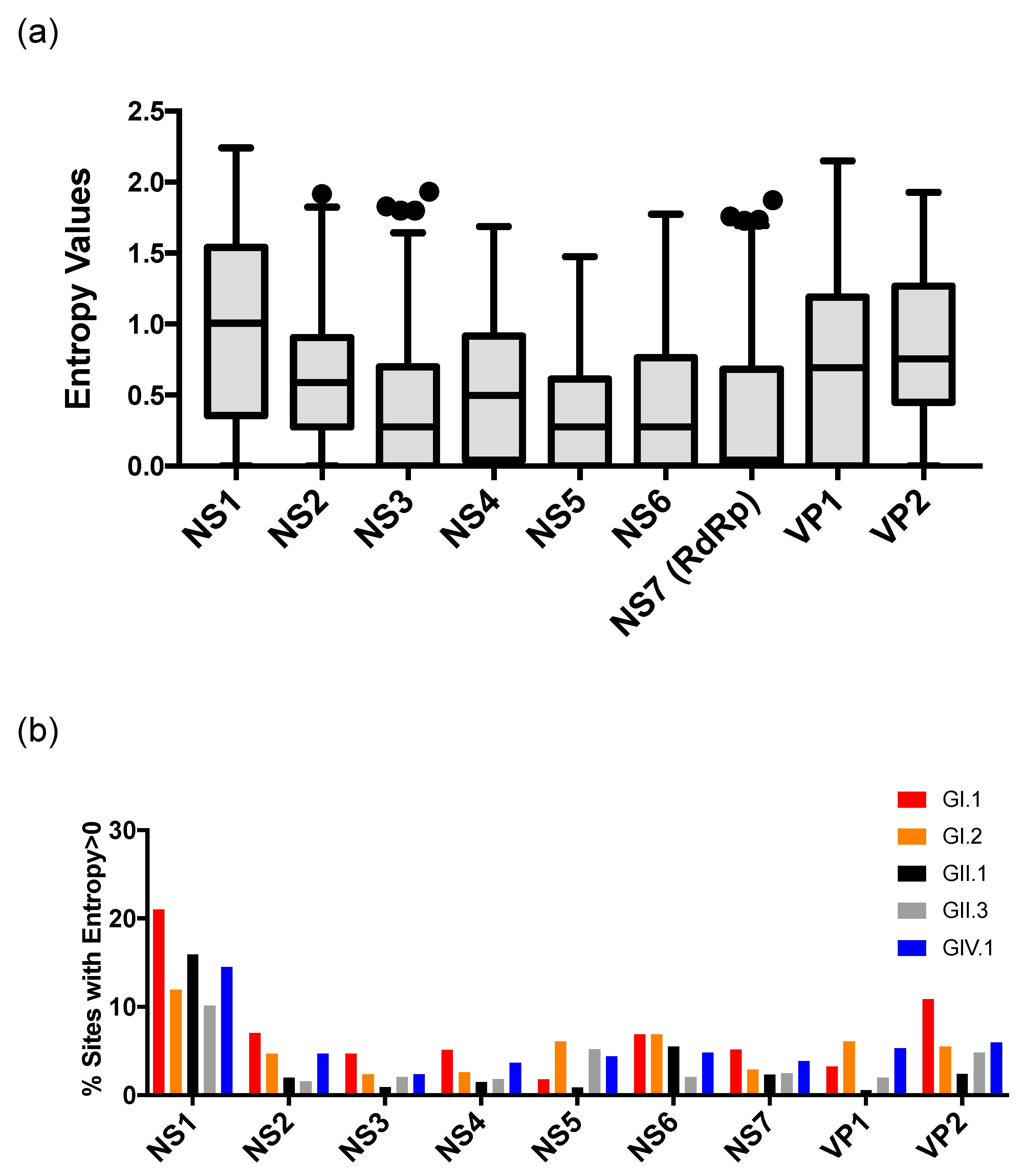
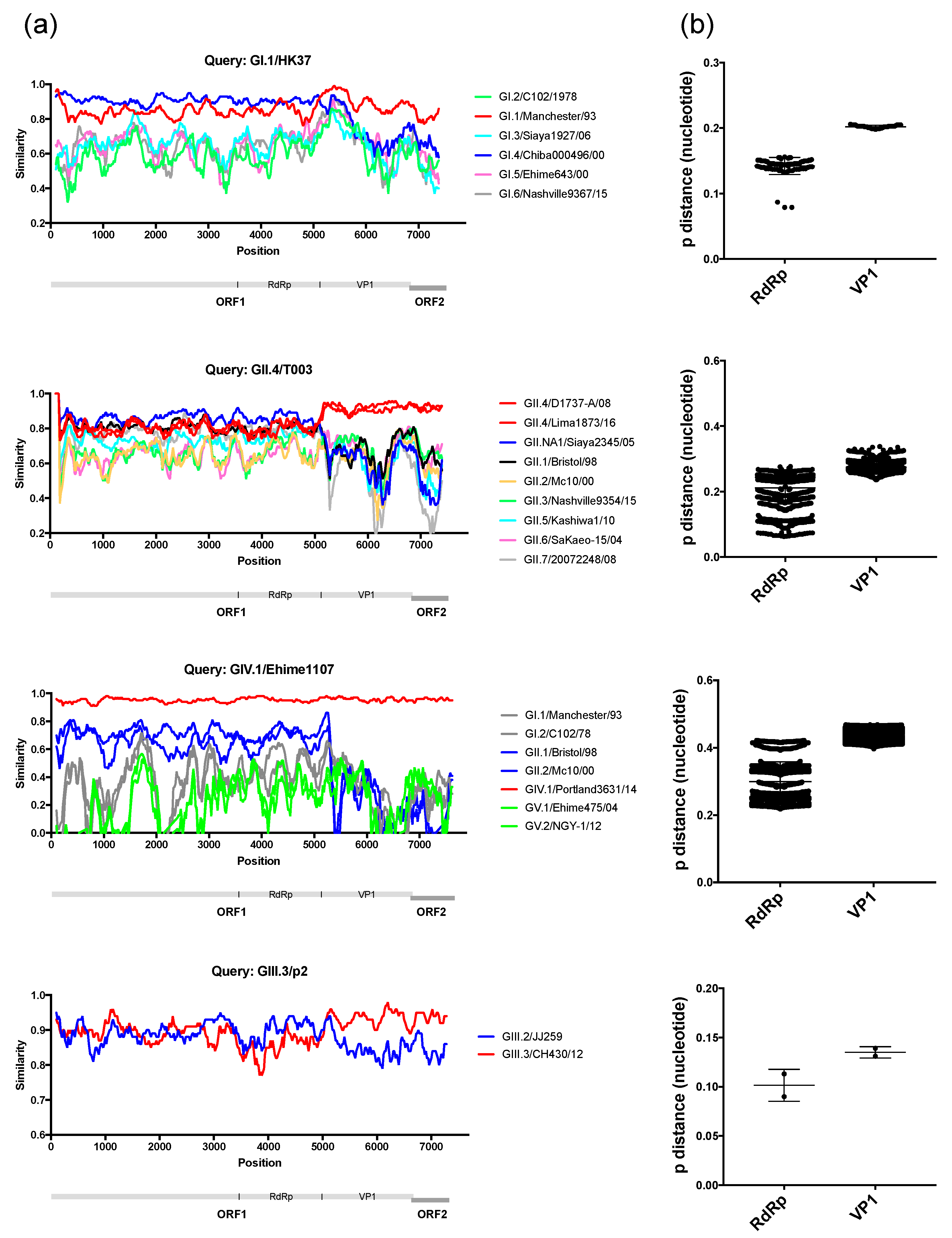
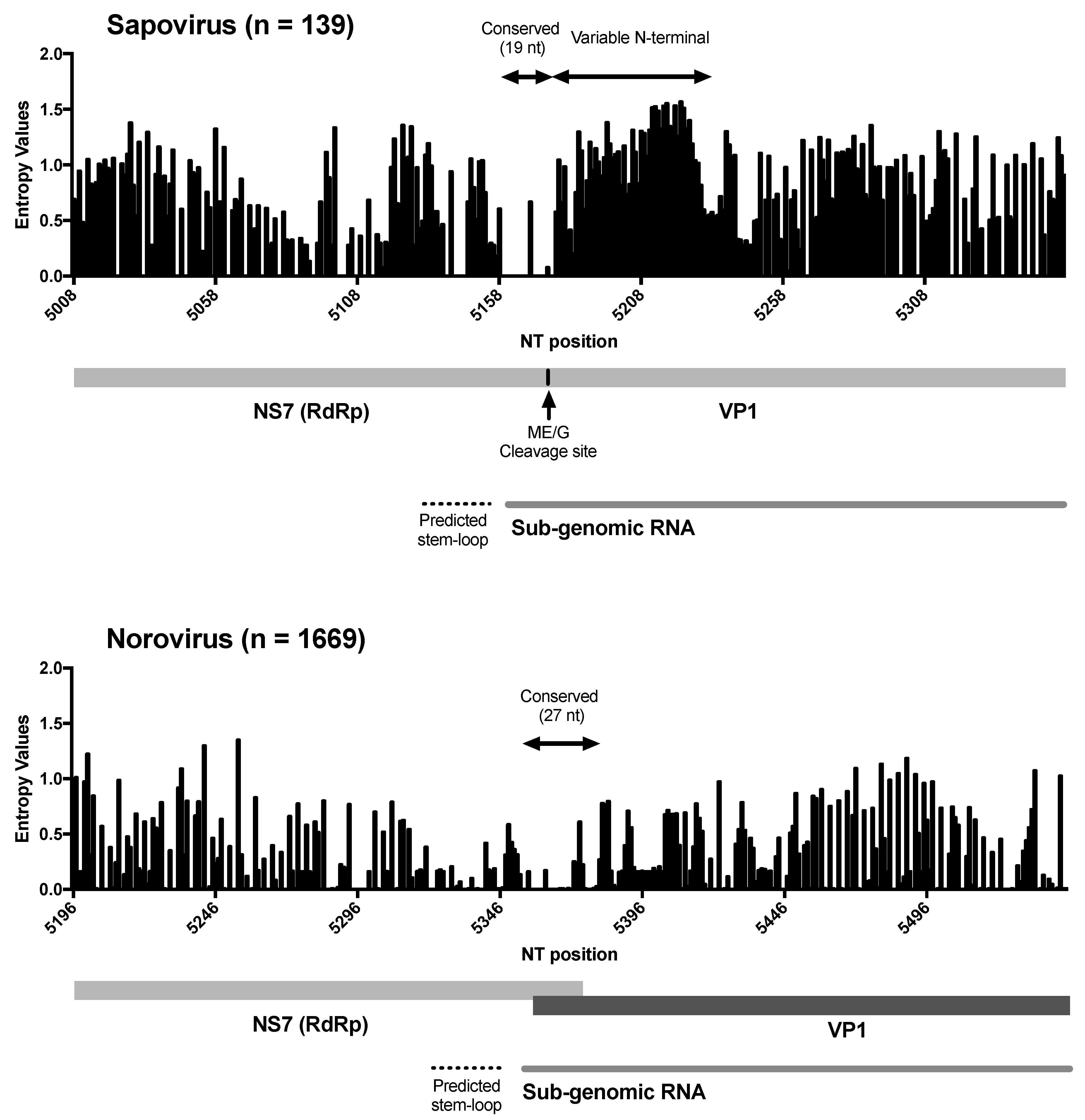
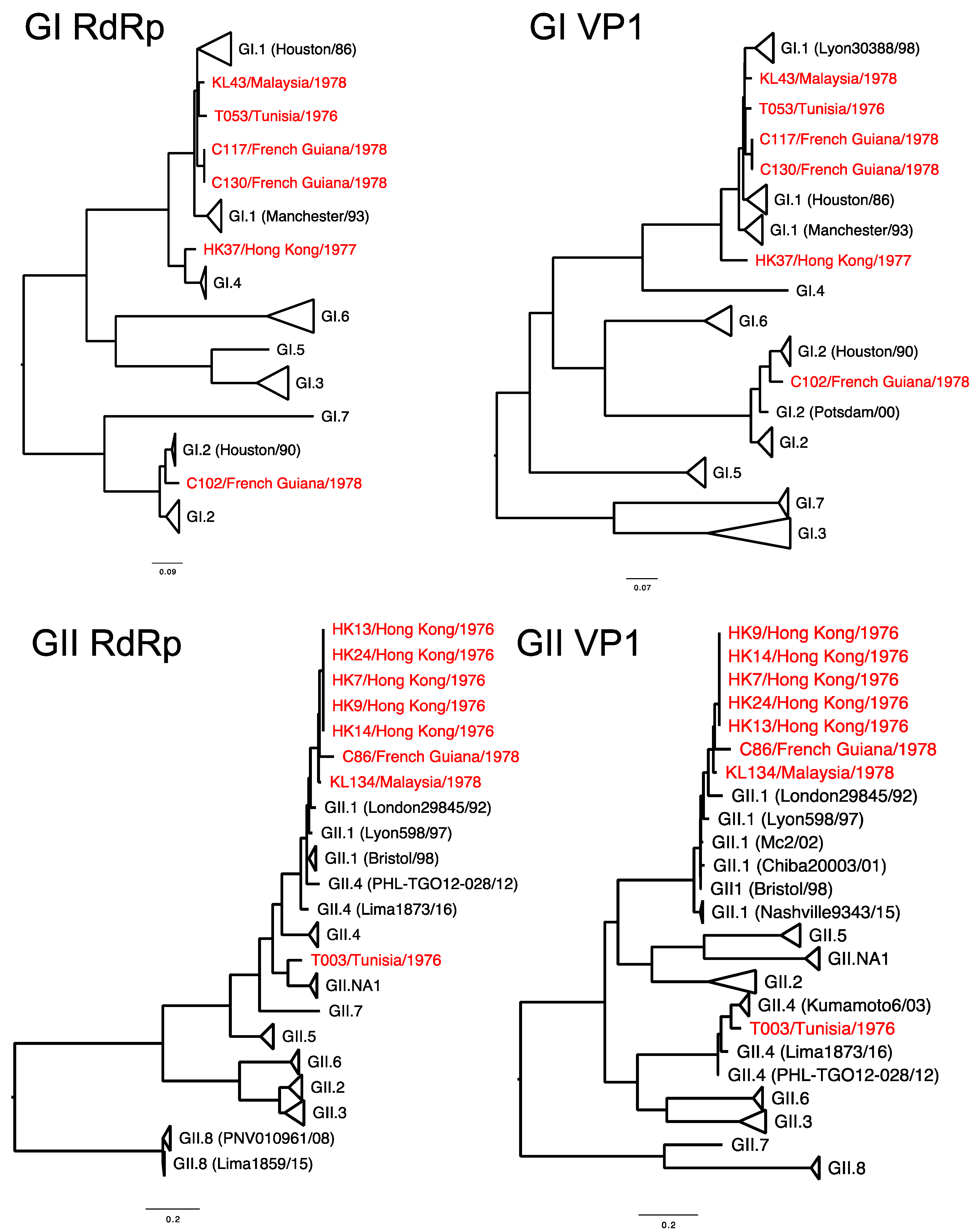
2.3. Sequence Analyses
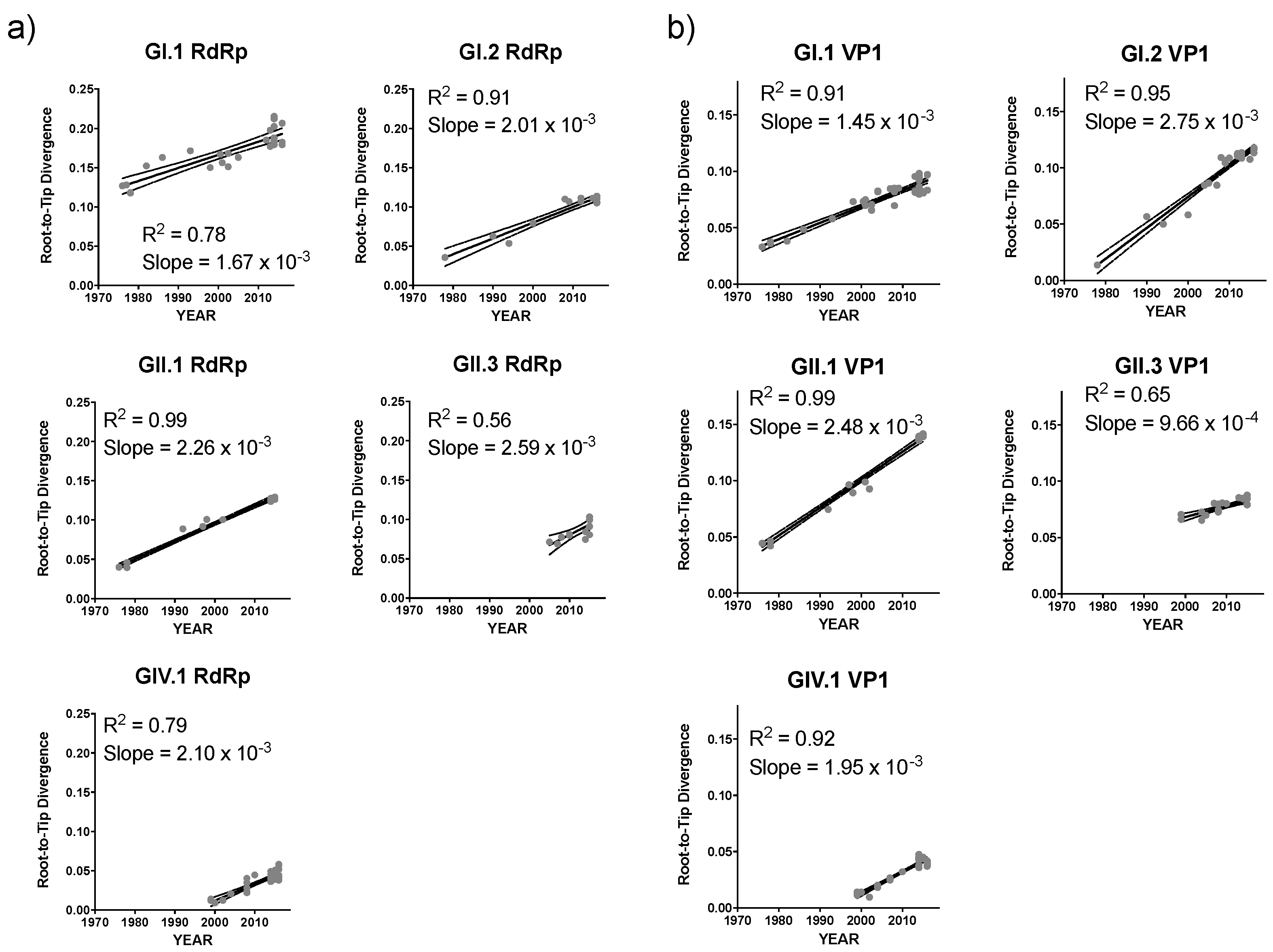
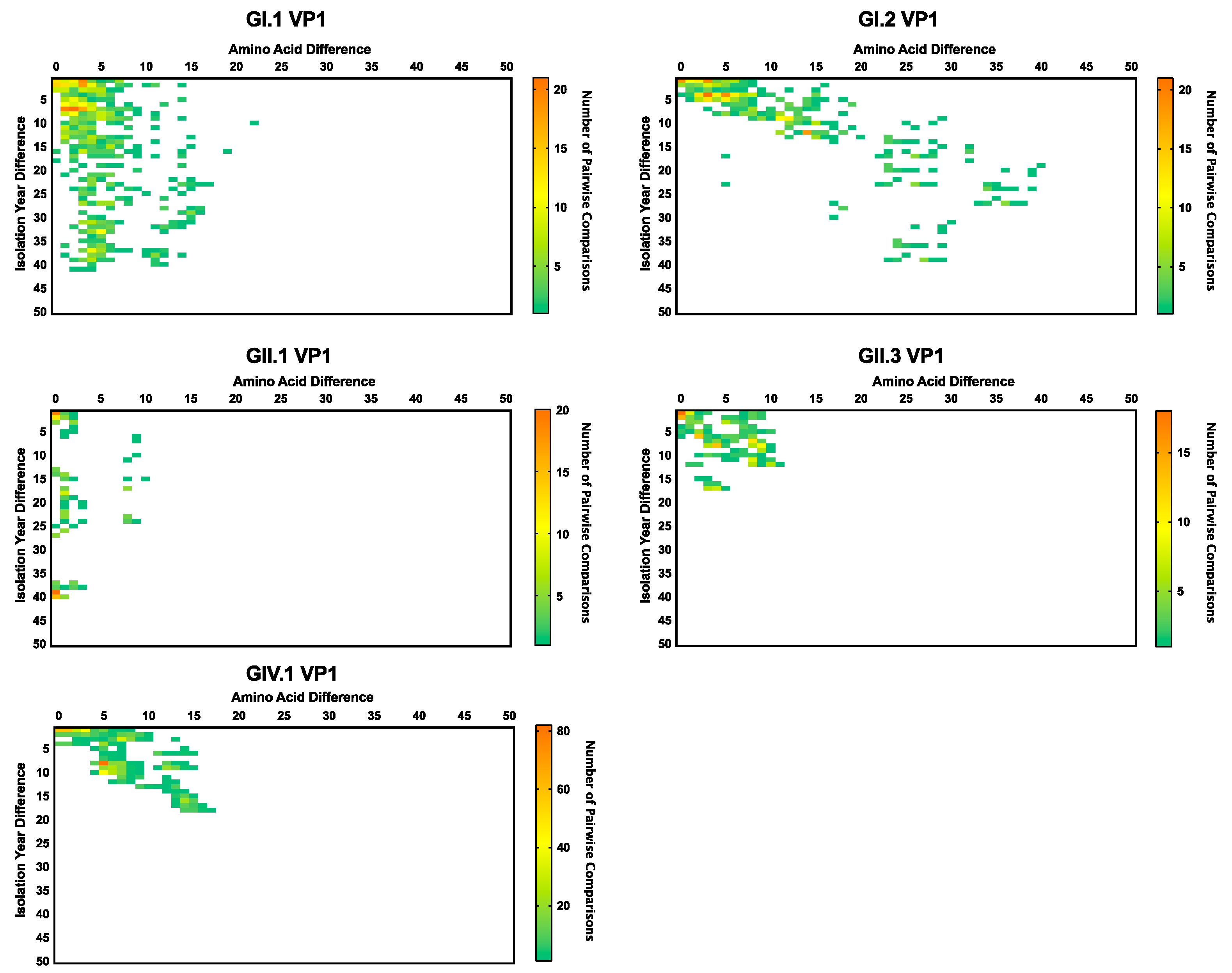
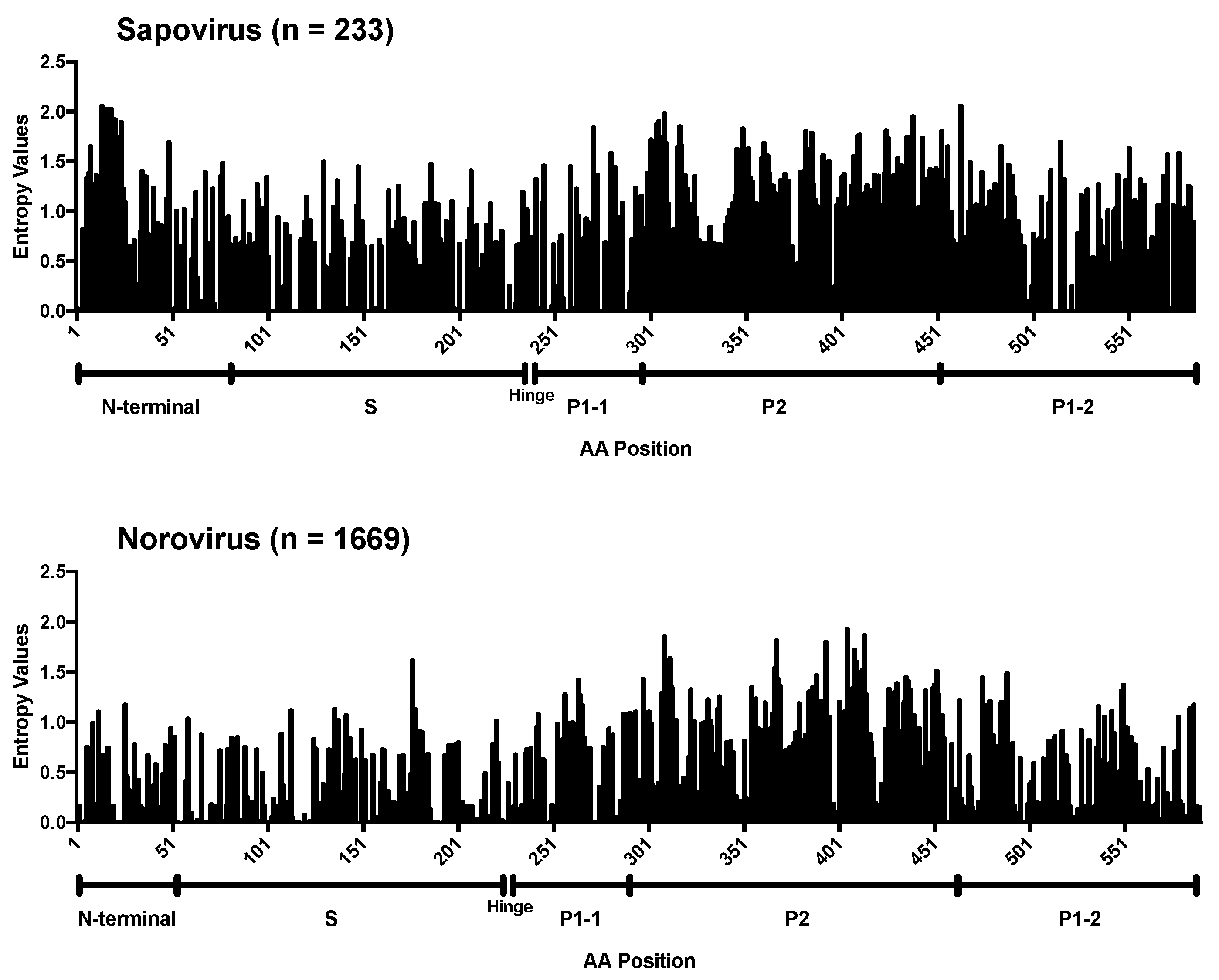
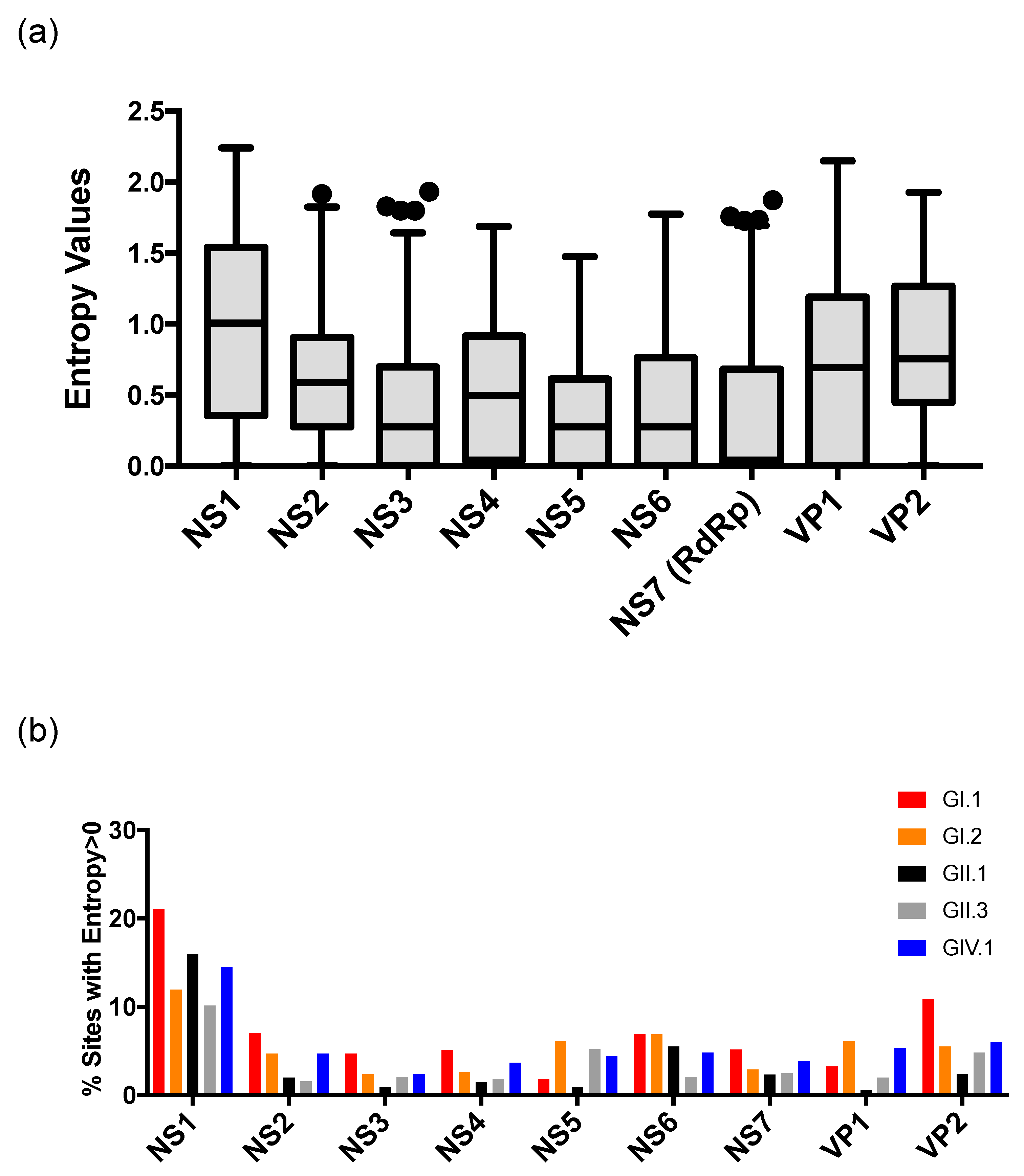
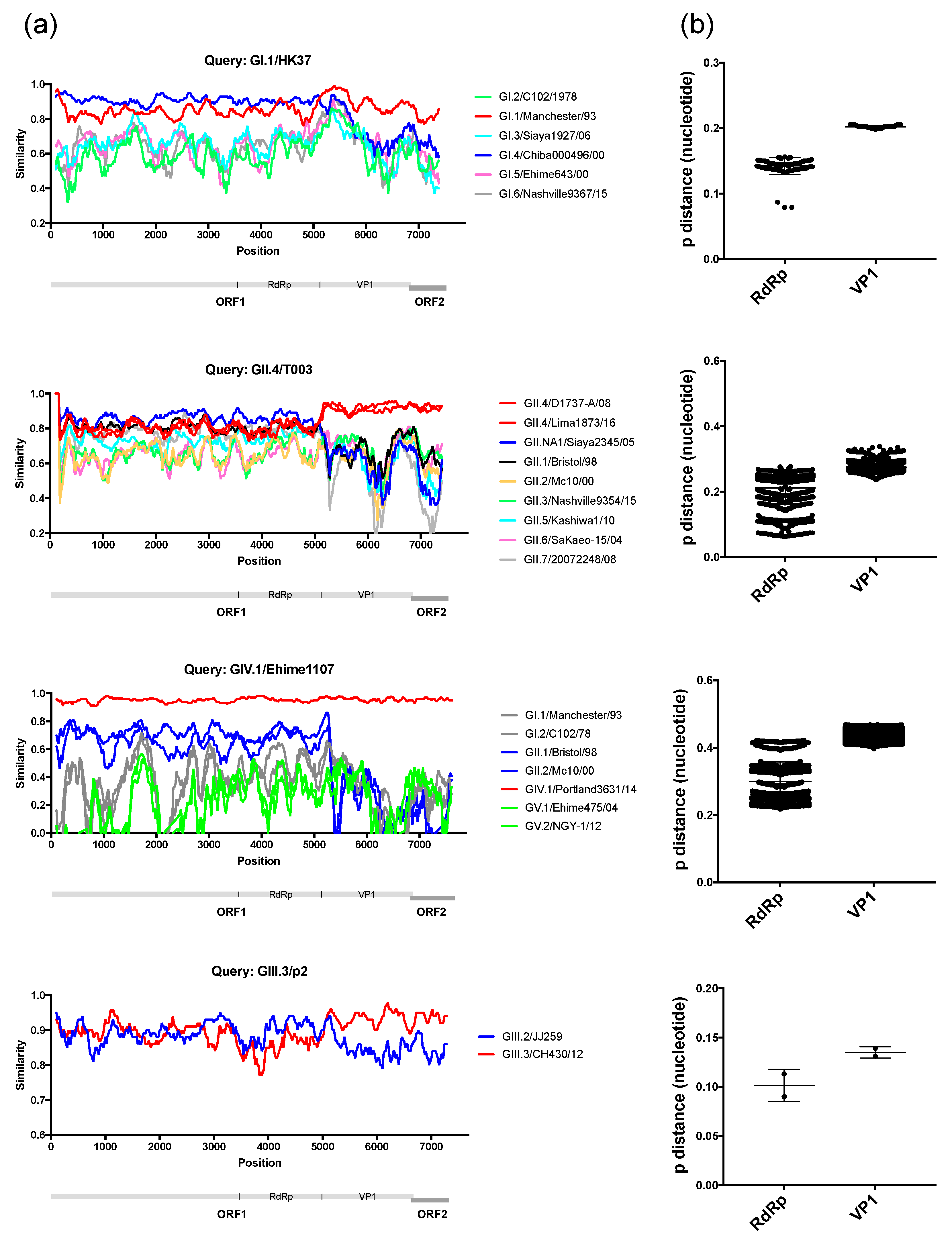
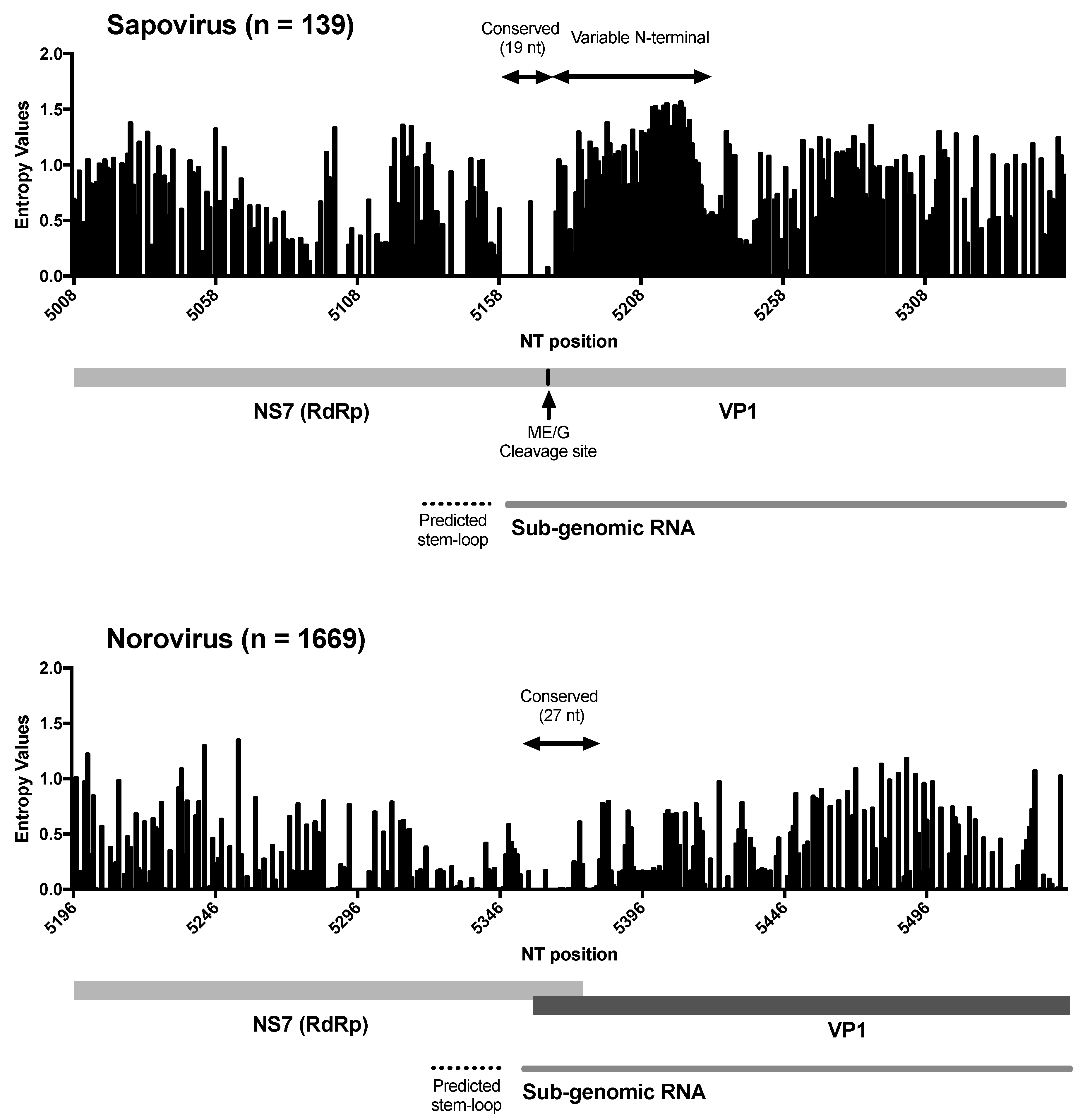
3. Results and Discussion
References.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oka, T.; Wang, Q.; Katayama, K.; Saif, L.J. Comprehensive review of human sapoviruses. Clin. Microbiol. Rev. 2015, 28, 32–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, I.N.; Lambden, P.R. Organization and expression of calicivirus genes. J. Infect. Dis. 2000, 181 (Suppl. 2), S309–S316. [Google Scholar] [CrossRef]
- Miyazaki, N.; Taylor, D.W.; Hansman, G.S.; Murata, K. Antigenic and Cryo-Electron Microscopy Structure Analysis of a Chimeric Sapovirus Capsid. J. Virol. 2015, 90, 2664–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Mori, K.; Iritani, N.; Harada, S.; Ueki, Y.; Iizuka, S.; Mise, K.; Murakami, K.; Wakita, T.; Katayama, K. Human sapovirus classification based on complete capsid nucleotide sequences. Arch. Virol. 2012, 157, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Diez-Valcarce, M.; Castro, C.J.; Marine, R.L.; Halasa, N.; Mayta, H.; Saito, M.; Tsaknaridis, L.; Pan, C.Y.; Bucardo, F.; Becker-Dreps, S.; et al. Genetic diversity of human sapovirus across the Americas. J. Clin. Virol. 2018, 104, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Hansman, G.S.; Takeda, N.; Oka, T.; Oseto, M.; Hedlund, K.O.; Katayama, K. Intergenogroup recombination in sapoviruses. Emerg. Infect. Dis. 2005, 11, 1916–1920. [Google Scholar] [CrossRef]
- Katayama, K.; Miyoshi, T.; Uchino, K.; Oka, T.; Tanaka, T.; Takeda, N.; Hansman, G.S. Novel recombinant sapovirus. Emerg. Infect. Dis. 2004, 10, 1874–1876. [Google Scholar] [CrossRef]
- Kuroda, M.; Masuda, T.; Ito, M.; Naoi, Y.; Doan, Y.H.; Haga, K.; Tsuchiaka, S.; Kishimoto, M.; Sano, K.; Omatsu, T.; et al. Genetic diversity and intergenogroup recombination events of sapoviruses detected from feces of pigs in Japan. Infect. Genet. Evol. 2017, 55, 209–217. [Google Scholar] [CrossRef]
- Li, J.; Shen, Q.; Zhang, W.; Zhao, T.; Li, Y.; Jiang, J.; Yu, X.; Guo, Z.; Cui, L.; Hua, X. Genomic organization and recombination analysis of a porcine sapovirus identified from a piglet with diarrhea in China. Virol. J. 2017, 14, 57. [Google Scholar] [CrossRef] [Green Version]
- Kumthip, K.; Khamrin, P.; Ushijima, H.; Chen, L.; Li, S.; Maneekarn, N. Genetic recombination and diversity of sapovirus in pediatric patients with acute gastroenteritis in Thailand, 2010–2018. PeerJ 2020, 8, e8520. [Google Scholar] [CrossRef]
- Liu, X.; Yamamoto, D.; Saito, M.; Imagawa, T.; Ablola, A.; Tandoc, A.O., 3rd; Segubre-Mercado, E.; Lupisan, S.P.; Okamoto, M.; Furuse, Y.; et al. Molecular detection and characterization of sapovirus in hospitalized children with acute gastroenteritis in the Philippines. J. Clin. Virol. 2015, 68, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Anjos, K.; Lima, L.M.; Silva, P.A.; Inoue-Nagata, A.K.; Nagata, T. The possible molecular evolution of sapoviruses by inter- and intra-genogroup recombination. Arch. Virol. 2011, 156, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Han, M.G.; Funk, J.A.; Bowman, G.; Janies, D.A.; Saif, L.J. Genetic diversity and recombination of porcine sapoviruses. J. Clin. Microbiol. 2005, 43, 5963–5972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, R.A.; Tanaka, M.M.; White, P.A. Norovirus recombination. J. Gen. Virol. 2007, 88, 3347–3359. [Google Scholar] [CrossRef] [PubMed]
- Desselberger, U. Caliciviridae Other Than Noroviruses. Viruses 2019, 11, 286. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, C.; Liebert, U.G. Intrahost viral evolution during chronic sapovirus infections. J. Clin. Virol. 2019, 113, 1–7. [Google Scholar] [CrossRef]
- Hergens, M.P.; Nederby Ohd, J.; Alm, E.; Askling, H.H.; Helgesson, S.; Insulander, M.; Lagerqvist, N.; Svenungsson, B.; Tihane, M.; Tolfvenstam, T.; et al. Investigation of a food-borne outbreak of gastroenteritis in a school canteen revealed a variant of sapovirus genogroup V not detected by standard PCR, Sollentuna, Sweden, 2016. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef]
- Pang, X.L.; Lee, B.E.; Tyrrell, G.J.; Preiksaitis, J.K. Epidemiology and genotype analysis of sapovirus associated with gastroenteritis outbreaks in Alberta, Canada: 2004–2007. J. Infect. Dis. 2009, 199, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.J.; Bergentoft, K.; Larsson, P.A.; Magnusson, G.; Widell, A.; Thorhagen, M.; Hedlund, K.O. A nosocomial sapovirus-associated outbreak of gastroenteritis in adults. Scand. J. Infect. Dis. 2005, 37, 200–204. [Google Scholar] [CrossRef]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.; Donnelly, C.A.; Cauchemez, S.; Hanage, W.P.; Van Kerkhove, M.D.; Hollingsworth, T.D.; Griffin, J.; Baggaley, R.F.; Jenkins, H.E.; Lyons, E.J.; et al. Pandemic potential of a strain of influenza A (H1N1): Early findings. Science 2009, 324, 1557–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, G.L.; MacCannell, D.R.; Taylor, J.; Carleton, H.A.; Neuhaus, E.B.; Bradbury, R.S.; Posey, J.E.; Gwinn, M. Pathogen Genomics in Public Health. N. Engl. J. Med. 2019, 381, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, M.; MacCannell, D.; Armstrong, G.L. Next-Generation Sequencing of Infectious Pathogens. JAMA 2019, 321, 893–894. [Google Scholar] [CrossRef] [Green Version]
- Biek, R.; Pybus, O.G.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Kao, R.R.; Haydon, D.T.; Lycett, S.J.; Murcia, P.R. Supersize me: How whole-genome sequencing and big data are transforming epidemiology. Trends Microbiol. 2014, 22, 282–291. [Google Scholar] [CrossRef]
- Timme, R.E.; Strain, E.; Baugher, J.D.; Davis, S.; Gonzalez-Escalona, N.; Sanchez Leon, M.; Allard, M.W.; Brown, E.W.; Tallent, S.; Rand, H. Phylogenomic Pipeline Validation for Foodborne Pathogen Disease Surveillance. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [Green Version]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Giombini, E.; Rozera, G. Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013, 19, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Roy, S.; Ruis, C.; Yara Romero, E.; Shah, D.; Williams, R.; Breuer, J. Norovirus Whole-Genome Sequencing by SureSelect Target Enrichment: A Robust and Sensitive Method. J. Clin. Microbiol. 2016, 54, 2530–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Leonard, S.R.; Mammel, M.K.; Elkins, C.A.; Kulka, M. Towards next-generation sequencing analytics for foodborne RNA viruses: Examining the effect of RNA input quantity and viral RNA purity. J. Virol. Methods 2016, 236, 221–230. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Allergy and Infectious Diseases. Annual Report of Program Activities; Government Printing Office: Washington, DC, USA, 1979. [Google Scholar]
- Katayama, K.; Shirato-Horikoshi, H.; Kojima, S.; Kageyama, T.; Oka, T.; Hoshino, F.; Fukushi, S.; Shinohara, M.; Uchida, K.; Suzuki, Y.; et al. Phylogenetic analysis of the complete genome of 18 Norwalk-like viruses. Virology 2002, 299, 225–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef]
- Simonyan, V.; Chumakov, K.; Dingerdissen, H.; Faison, W.; Goldweber, S.; Golikov, A.; Gulzar, N.; Karagiannis, K.; Vinh Nguyen Lam, P.; Maudru, T.; et al. High-performance integrated virtual environment (HIVE): A robust infrastructure for next-generation sequence data analysis. Database (Oxford) 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Tohma, K.; Saito, M.; Mayta, H.; Zimic, M.; Lepore, C.J.; Ford-Siltz, L.A.; Gilman, R.H.; Parra, G.I. Complete Genome Sequence of a Nontypeable GII Norovirus Detected in Peru. Genome Announc. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Evolutionary dynamics of non-GII genotype 4 (GII.4) noroviruses reveal limited and independent diversification of variants. J. Gen. Virol. 2018, 99, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.F.; Duraes-Carvalho, R.; Oliveira-Filho, E.F.; Alfieri, A.A.; Van der Poel, W.H.M. High-resolution phylogeny providing insights towards the epidemiology, zoonotic aspects and taxonomy of sapoviruses. Infect. Genet. Evol. 2017, 56, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Yamamoto, M.; Katayama, K.; Hansman, G.S.; Ogawa, S.; Miyamura, T.; Takeda, N. Identification of the cleavage sites of sapovirus open reading frame 1 polyprotein. J. Gen. Virol. 2006, 87, 3329–3338. [Google Scholar] [CrossRef]
- Okada, M.; Yamashita, Y.; Oseto, M.; Ogawa, T.; Kaiho, I.; Shinozaki, K. Genetic variability in the sapovirus capsid protein. Virus Genes 2006, 33, 157–161. [Google Scholar] [CrossRef]
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Gallimore, C.I.; Iturriza-Gomara, M.; Lewis, D.; Cubitt, D.; Cotterill, H.; Gray, J.J. Characterization of sapoviruses collected in the United Kingdom from 1989 to 2004. J. Med. Virol. 2006, 78, 673–682. [Google Scholar] [CrossRef]
- Johnsen, C.K.; Midgley, S.; Bottiger, B. Genetic diversity of sapovirus infections in Danish children 2005–2007. J. Clin. Virol. 2009, 46, 265–269. [Google Scholar] [CrossRef]
- Kumthip, K.; Khamrin, P.; Maneekarn, N. Molecular epidemiology and genotype distributions of noroviruses and sapoviruses in Thailand 2000-2016: A review. J. Med. Virol. 2018, 90, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Lasure, N.; Gopalkrishna, V. Epidemiological profile and genetic diversity of sapoviruses (SaVs) identified in children suffering from acute gastroenteritis in Pune, Maharashtra, Western India, 2007-2011. Epidemiol. Infect. 2017, 145, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, P.; Pietsch, C.; Liebert, U.G. Genetic Diversity of Sapoviruses among Inpatients in Germany, 2008–2018. Viruses 2019, 11, 726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, T.Y.; Nadan, S.; Page, N.A.; Taylor, M.B. Diverse sapovirus genotypes identified in children hospitalised with gastroenteritis in selected regions of South Africa. J. Clin. Virol. 2016, 76, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, G.J.; Mayta, H.; Pajuelo, M.J.; Neira, K.; Xiaofang, L.; Cabrera, L.; Ballard, S.B.; Crabtree, J.E.; Kelleher, D.; Cama, V.; et al. Epidemiology of Sapovirus Infections in a Birth Cohort in Peru. Clin. Infect. Dis. 2018, 66, 1858–1863. [Google Scholar] [CrossRef] [Green Version]
- Svraka, S.; Vennema, H.; van der Veer, B.; Hedlund, K.O.; Thorhagen, M.; Siebenga, J.; Duizer, E.; Koopmans, M. Epidemiology and genotype analysis of emerging sapovirus-associated infections across Europe. J. Clin. Microbiol. 2010, 48, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Platts-Mills, J.A.; Liu, J.; Rogawski, E.T.; Kabir, F.; Lertsethtakarn, P.; Siguas, M.; Khan, S.S.; Praharaj, I.; Murei, A.; Nshama, R.; et al. Use of quantitative molecular diagnostic methods to assess the aetiology, burden, and clinical characteristics of diarrhoea in children in low-resource settings: A reanalysis of the MAL-ED cohort study. Lancet Glob. Health 2018, 6, e1309–e1318. [Google Scholar] [CrossRef] [Green Version]
- Grant, L.R.; O’Brien, K.L.; Weatherholtz, R.C.; Reid, R.; Goklish, N.; Santosham, M.; Parashar, U.; Vinje, J. Norovirus and Sapovirus Epidemiology and Strain Characteristics among Navajo and Apache Infants. PLoS ONE 2017, 12, e0169491. [Google Scholar] [CrossRef]
- Hassan, F.; Kanwar, N.; Harrison, C.J.; Halasa, N.B.; Chappell, J.D.; Englund, J.A.; Klein, E.J.; Weinberg, G.A.; Szilagyi, P.G.; Moffatt, M.E.; et al. Viral Etiology of Acute Gastroenteritis in <2-Year-Old US Children in the Post-Rotavirus Vaccine Era. J. Pediatric Infect. Dis. Soc. 2019, 8, 414–421. [Google Scholar] [CrossRef]
- Torner, N.; Martinez, A.; Broner, S.; Moreno, A.; Camps, N.; Dominguez, A.; Working Group for the Study of Acute Viral Gastroenteritis Outbreaks in Catalonia. Epidemiology of Acute Gastroenteritis Outbreaks Caused by Human Calicivirus (Norovirus and Sapovirus) in Catalonia: A Two Year Prospective Study, 2010–2011. PLoS ONE 2016, 11, e0152503. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C. Different rates of substitution may produce different phylogenies of the eutherian mammals. J. Mol. Evol. 1991, 33, 209–215. [Google Scholar] [CrossRef]
- Sun, S.; Evans, B.J.; Golding, G.B. “Patchy-tachy” leads to false positives for recombination. Mol. Biol. Evol. 2011, 28, 2549–2559. [Google Scholar] [CrossRef] [Green Version]
- Worobey, M.; Rambaut, A.; Pybus, O.G.; Robertson, D.L. Questioning the evidence for genetic recombination in the 1918 “Spanish flu” virus. Science 2002, 296. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, Y.J.; Johansson, M.; Norberg, P. Revisiting Recombination Signal in the Tick-Borne Encephalitis Virus: A Simulation Approach. PLoS ONE 2016, 11, e0164435. [Google Scholar] [CrossRef]
- Parra, G.I. Emergence of norovirus strains: A tale of two genes. Virus Evol. 2019, 5. [Google Scholar] [CrossRef]
- Bull, R.A.; Hansman, G.S.; Clancy, L.E.; Tanaka, M.M.; Rawlinson, W.D.; White, P.A. Norovirus recombination in ORF1/ORF2 overlap. Emerg. Infect. Dis. 2005, 11, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Kim, Y.; Saif, L.J.; Green, K.Y. Bile acids are essential for porcine enteric calicivirus replication in association with down-regulation of signal transducer and activator of transcription 1. Proc. Natl. Acad. Sci. USA 2004, 101, 8733–8738. [Google Scholar] [CrossRef] [Green Version]
- Bentley, K.; Evans, D.J. Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Perez-Losada, M.; Arenas, M.; Galan, J.C.; Palero, F.; Gonzalez-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.Z.; Worobey, M. Homologous recombination in negative sense RNA viruses. Viruses 2011, 3, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Taucher, C.; Berger, A.; Mandl, C.W. A trans-complementing recombination trap demonstrates a low propensity of flaviviruses for intermolecular recombination. J. Virol. 2010, 84, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P.; Karakasiliotis, I.; Bailey, D.; Chaudhry, Y.; Evans, D.J.; Goodfellow, I.G. Bioinformatic and functional analysis of RNA secondary structure elements among different genera of human and animal caliciviruses. Nucleic Acids Res. 2008, 36, 2530–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timme, R.E.; Sanchez Leon, M.; Allard, M.W. Utilizing the Public GenomeTrakr Database for Foodborne Pathogen Traceback. Methods Mol. Biol. 2019, 1918, 201–212. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | GI.1 | GI.2 | GI.3 | GI.4 | GI.5 | GI.6 | GI.7 | GII.1 | GII.2 | GII.3 | GII.4 | GII.5 | GII.6 | GII.7 | GII.8 | GII.NA1 | GIV.1 | GV.1 | GV.2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1976 | 1 * | 5 * | 1 * | ||||||||||||||||
| 1977 | 1 * | ||||||||||||||||||
| 1978 | 3 * | 1 * | 2 * | ||||||||||||||||
| 1982 | 2 | ||||||||||||||||||
| 1986 | 1 | ||||||||||||||||||
| 1990 | 1 | 1 | |||||||||||||||||
| 1992 | 1 | ||||||||||||||||||
| 1993 | 1 | ||||||||||||||||||
| 1994 | 1 | ||||||||||||||||||
| 1995 | 1 | ||||||||||||||||||
| 1997 | 1 | ||||||||||||||||||
| 1998 | 1 | 1 | |||||||||||||||||
| 1999 | 1 | 2 | 3 | ||||||||||||||||
| 2000 | 1 | 1 | 2 | 1 | 1 | 1 | 2 | ||||||||||||
| 2000/01 | 1 | ||||||||||||||||||
| 2001 | 2 | 1 | 1 | 1 | |||||||||||||||
| 2002 | 1 | 1 | |||||||||||||||||
| 2002/03 | 2 | 1 | |||||||||||||||||
| 2003 | 1 | 1 | |||||||||||||||||
| 2004 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | |||||||||||
| 2005 | 1 | 3 | 1 | 2 | 2 | 1 | |||||||||||||
| 2006 | 1 | 1 | 1 | ||||||||||||||||
| 2007 | 1 | 1 | 1 | 13 | |||||||||||||||
| 2008 | 7 | 1 | 1 | 1 | 1 | 5 | 1 | 2 | 1 | 1 | 1 | 7 | |||||||
| 2009 | 1 | 2 | 1 | 2 | |||||||||||||||
| 2010 | 1 | 2 | 1 | 1 | 1 | ||||||||||||||
| 2011 | 2 | ||||||||||||||||||
| 2012 | 1 | 5 | 1 | 1 | 1 | ||||||||||||||
| 2013 | 4 | 8 | 1 | 1 | |||||||||||||||
| 2014 | 8 | 4 | 2 | 2 | 1 | 3 | 10 | 2 | |||||||||||
| 2015 | 2 | 1 | 1 | 4 | 1 | 7 | 3 | 1 | 5 | 5 | |||||||||
| 2016 | 3 | 10 | 1 | 1 | 1 | 3 | 7 | 2 | |||||||||||
| 2018 | 1 | ||||||||||||||||||
| Total | 45 | 37 | 3 | 3 | 3 | 5 | 2 | 21 | 9 | 26 | 7 | 7 | 3 | 1 | 10 | 2 | 51 | 13 | 3 |
| Genotype | NS1/NS2 | NS2/NS3 | NS3/NS4 | NS4/NS5 | NS5/NS6-NS7 | NS6-NS7/VP1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E/G | E/A | E/G | E/S | Q/G | Q/A | Q/G | Q/S | E/A | E/G | Q/A | E/A | E/G | E/S | ME/G | |
| GI.1 | 23 | 23 | 23 | 23 | 8 | 8 | 7 | 23 | |||||||
| GI.2 | 11 | 11 | 11 | 10 | 1 | 9 | 2 | 11 | |||||||
| GI.3 | 2 | 2 | 2 | 2 | 2 | 2 | |||||||||
| GI.4 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| GI.5 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| GI.6 | 3 | 3 | 3 | 3 | 3 | 3 | |||||||||
| GI.7 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| GII.1 | 17 | 17 | 17 | 17 | 17 | 17 | |||||||||
| GII.2 | 6 | 6 | 6 | 6 | 6 | 6 | |||||||||
| GII.3 | 12 | 12 | 12 | 12 | 11 | 12 | |||||||||
| GII.4 | 5 | 5 | 5 | 5 | 5 | 5 | |||||||||
| GII.5 | 6 | 6 | 6 | 6 | 6 | 6 | |||||||||
| GII.6 | 2 | 2 | 2 | 2 | 2 | 2 | |||||||||
| GII.7 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| GII.8 | 8 | 8 | 8 | 8 | 8 | 8 | |||||||||
| GII.NA1 | 2 | 2 | 2 | 2 | 2 | 2 | |||||||||
| GIV.1 | 27 | 27 | 27 | 27 | 27 | 27 | |||||||||
| GV.1 | 8 | 8 | 8 | 8 | 8 | 8 | |||||||||
| GV.2 | 3 | 3 | 3 | 3 | 3 | 3 | |||||||||
| Genotype | ORF1 | ORF2 | ||||||
|---|---|---|---|---|---|---|---|---|
| NS1 | NS2 | NS3 | NS4 | NS5 | NS6-NS7 | VP1 | VP2 | |
| GI.1 | 68 | 256 | 341 | 274 | 114 | 668 * | 559 | 166 |
| GI.2 | 67 | 256 | 341 | 274 | 115 | 668 | 569 | 164 |
| GI.3 | 68 | 256 | 341 | 274 | 114 | 668 | 564 | 166 |
| GI.4 | 68 | 256 | 341 | 274 | 114 | 668 | 559 | 166 |
| GI.5 | 68 | 256 | 341 | 274 | 114 | 668 | 565 | 166 |
| GI.6 | 67 | 256 | 341 | 274 | 114 | 668 | 563 | 166 |
| GI.7 | 67 | 256 | 341 | 274 | 115 | 668 | 566 | 166 |
| GII.1 | 69 | 256 | 341 | 274 | 115 | 667 | 558 | 167 |
| GII.2 | 69 | 256 | 341 | 274 | 115 | 667 | 556 | 167 |
| GII.3 | 69 | 256 | 341 | 274 | 115 | 667 | 559 | 167 |
| GII.4 | 69 | 256 | 341 | 274 | 115 | 667 | 557 | 167 |
| GII.5 | 69 | 256 | 341 | 274 | 115 | 667 | 557 | 167 |
| GII.6 | 69 | 256 | 341 | 274 | 115 | 667 | 559 | 167 |
| GII.7 | 69 | 256 | 341 | 274 | 115 | 667 | 556 | 167 |
| GII.8 | 70 | 256 | 341 | 275 | 115 | 667 | 554 | 167 |
| GII.NA1 | 69 | 256 | 341 | 274 | 115 | 667 | 557 | 167 |
| GIV.1 | 69 | 256 | 341 | 274 | 115 | 667 | 549 | 168 |
| GV.1 | 67 | 257 | 341 | 279 | 123 | 667 | 567 | 167 |
| GV.2 | 65 | 257 | 341 | 279 | 123 | 667 | 567 # | 168 |
| Genotype | Mean Substitution Rate, Subs/Site/Year (95% HPD Interval) | |
|---|---|---|
| RdRp | VP1 | |
| GI.1 | 2.25 (1.82–2.69) × 10−3 | 1.38 (1.14–1.65) × 10−3 |
| GI.2 | 3.38 (2.60–4.16) × 10−3 | 1.32 (0.95–1.70) × 10−3 |
| GII.1 | 2.90 (2.33–3.48) × 10−3 | 2.75 (2.21–3.29) × 10−3 |
| GII.3 | 2.82 (1.54–4.11) × 10−3 | 2.31 (1.69–2.94) × 10−3 |
| GIV.1 | 2.82 (2.29–3.36) × 10−3 | 2.00 (1.50–2.51) × 10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tohma, K.; Kulka, M.; Coughlan, S.; Green, K.Y.; Parra, G.I. Genomic Analyses of Human Sapoviruses Detected over a 40-Year Period Reveal Disparate Patterns of Evolution among Genotypes and Genome Regions. Viruses 2020, 12, 516. https://doi.org/10.3390/v12050516
Tohma K, Kulka M, Coughlan S, Green KY, Parra GI. Genomic Analyses of Human Sapoviruses Detected over a 40-Year Period Reveal Disparate Patterns of Evolution among Genotypes and Genome Regions. Viruses. 2020; 12(5):516. https://doi.org/10.3390/v12050516
Chicago/Turabian StyleTohma, Kentaro, Michael Kulka, Suzie Coughlan, Kim Y. Green, and Gabriel I. Parra. 2020. "Genomic Analyses of Human Sapoviruses Detected over a 40-Year Period Reveal Disparate Patterns of Evolution among Genotypes and Genome Regions" Viruses 12, no. 5: 516. https://doi.org/10.3390/v12050516
APA StyleTohma, K., Kulka, M., Coughlan, S., Green, K. Y., & Parra, G. I. (2020). Genomic Analyses of Human Sapoviruses Detected over a 40-Year Period Reveal Disparate Patterns of Evolution among Genotypes and Genome Regions. Viruses, 12(5), 516. https://doi.org/10.3390/v12050516