Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes

 ,
, 
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Data Acquisition
2.2. Nucleotide Composition Analysis
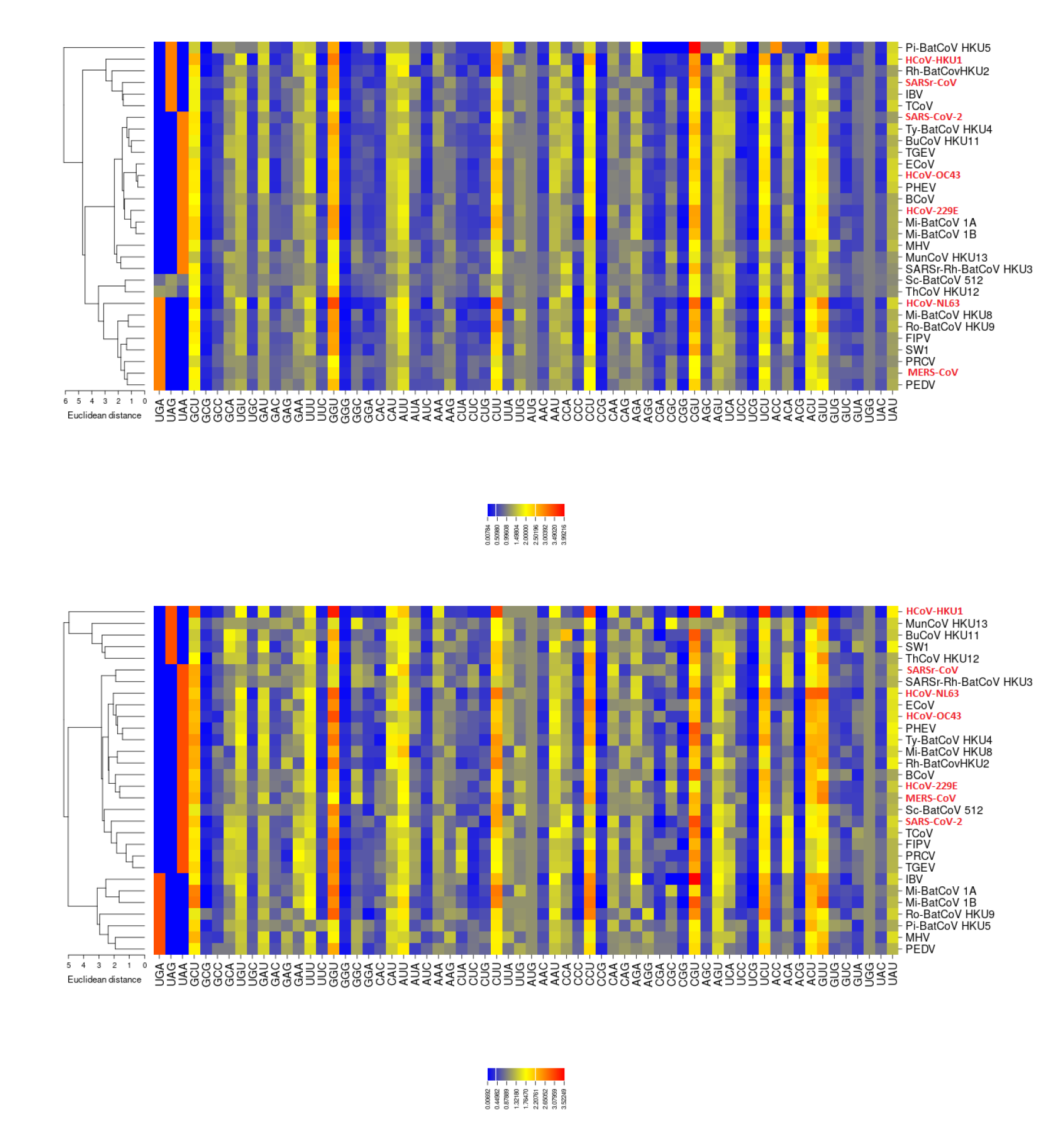
2.3. RSCU
2.4. Effective Number of Codons Analysis
2.5. Codon Adaptation Index
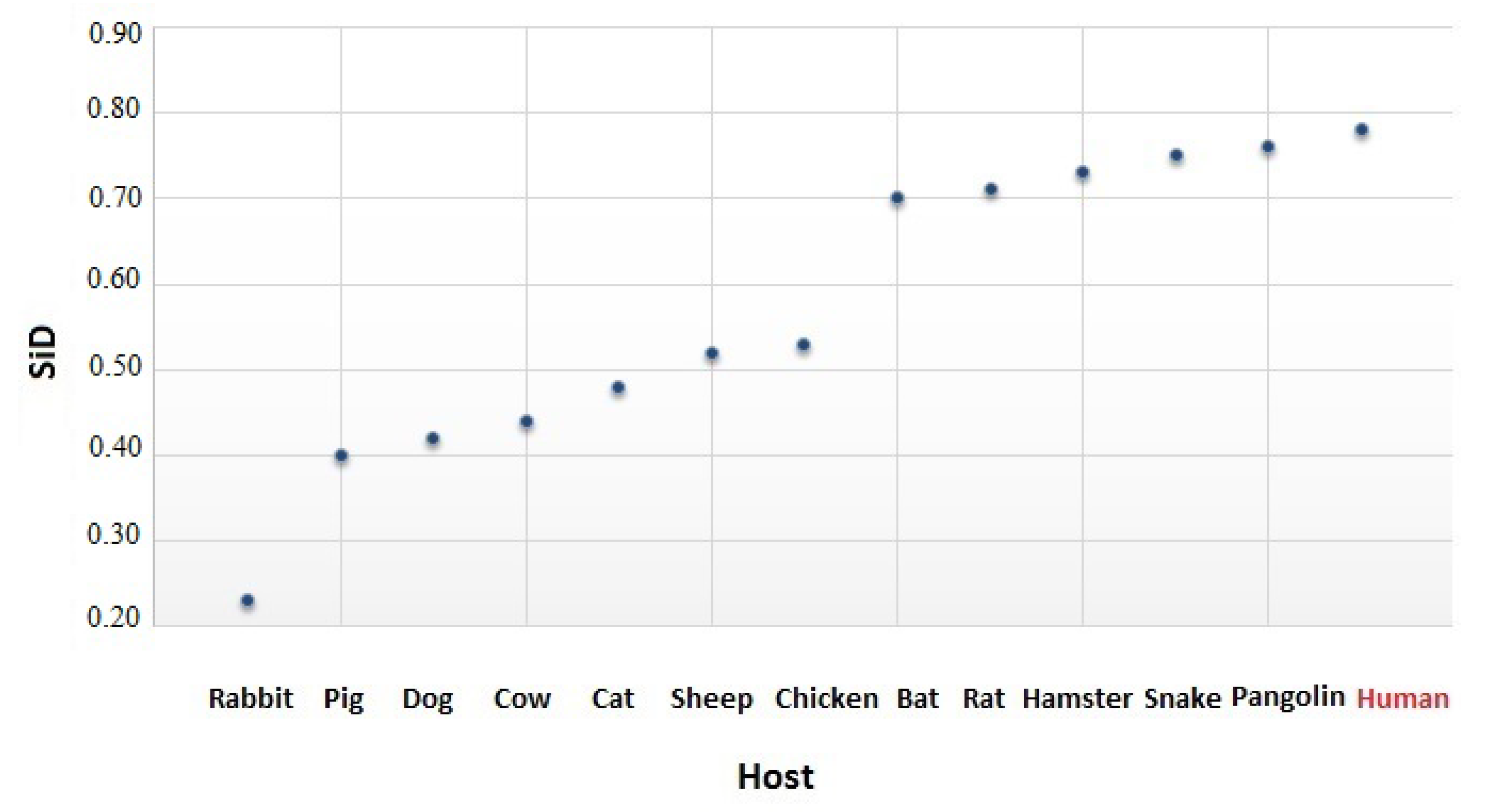
2.6. The Similarity Index
2.7. ENC Plot
2.8. Neutrality Plot
2.9. Forsdyke Plot
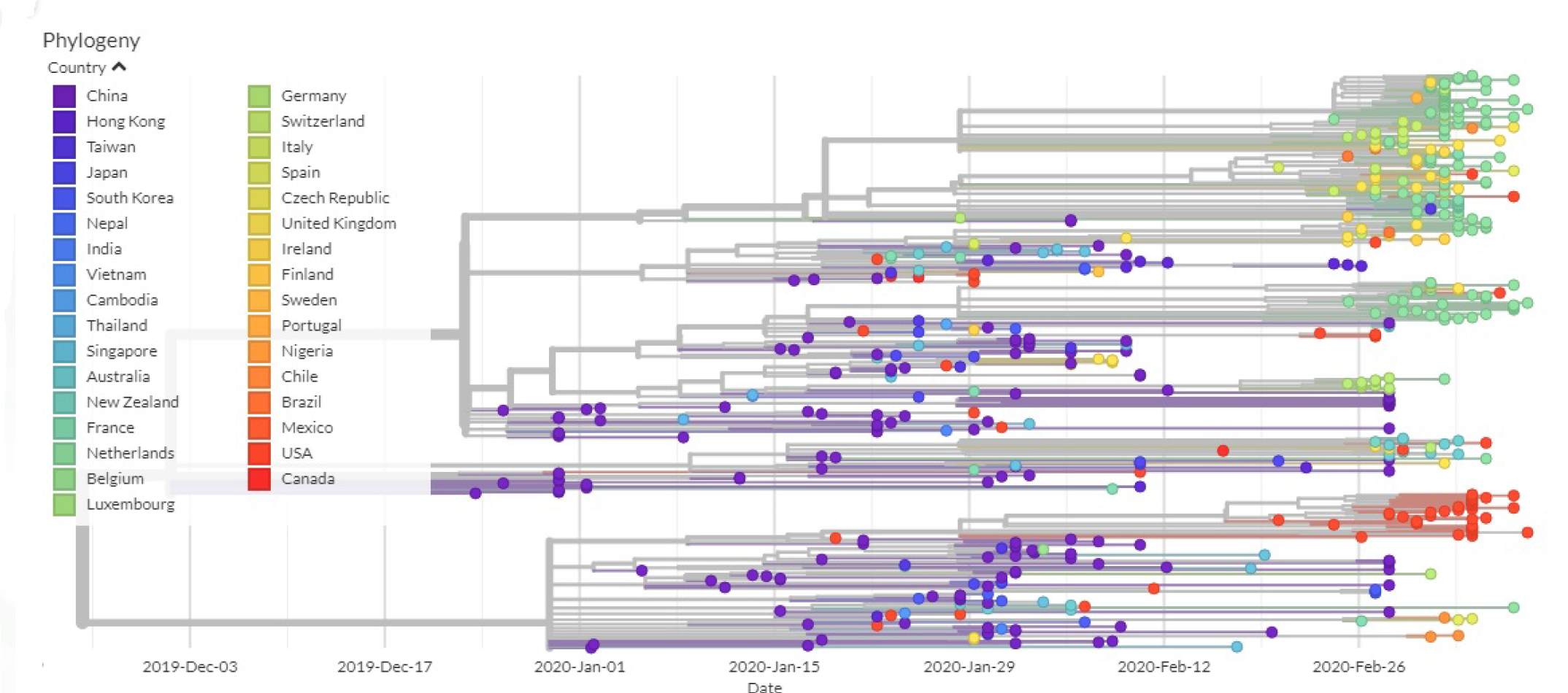
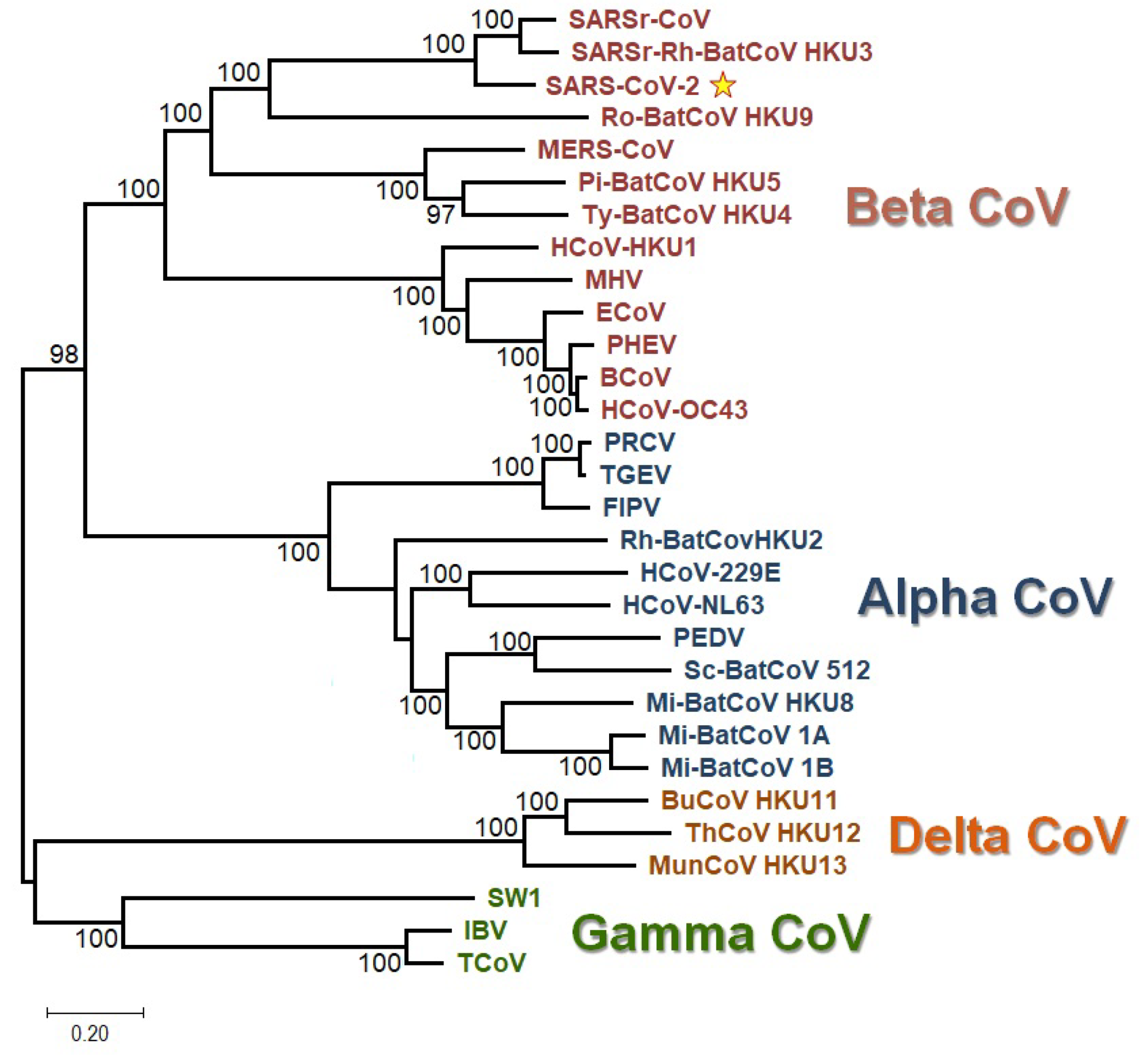
2.10. Phylogenetic Analysis
3. Results
3.1. Nucleotide Composition
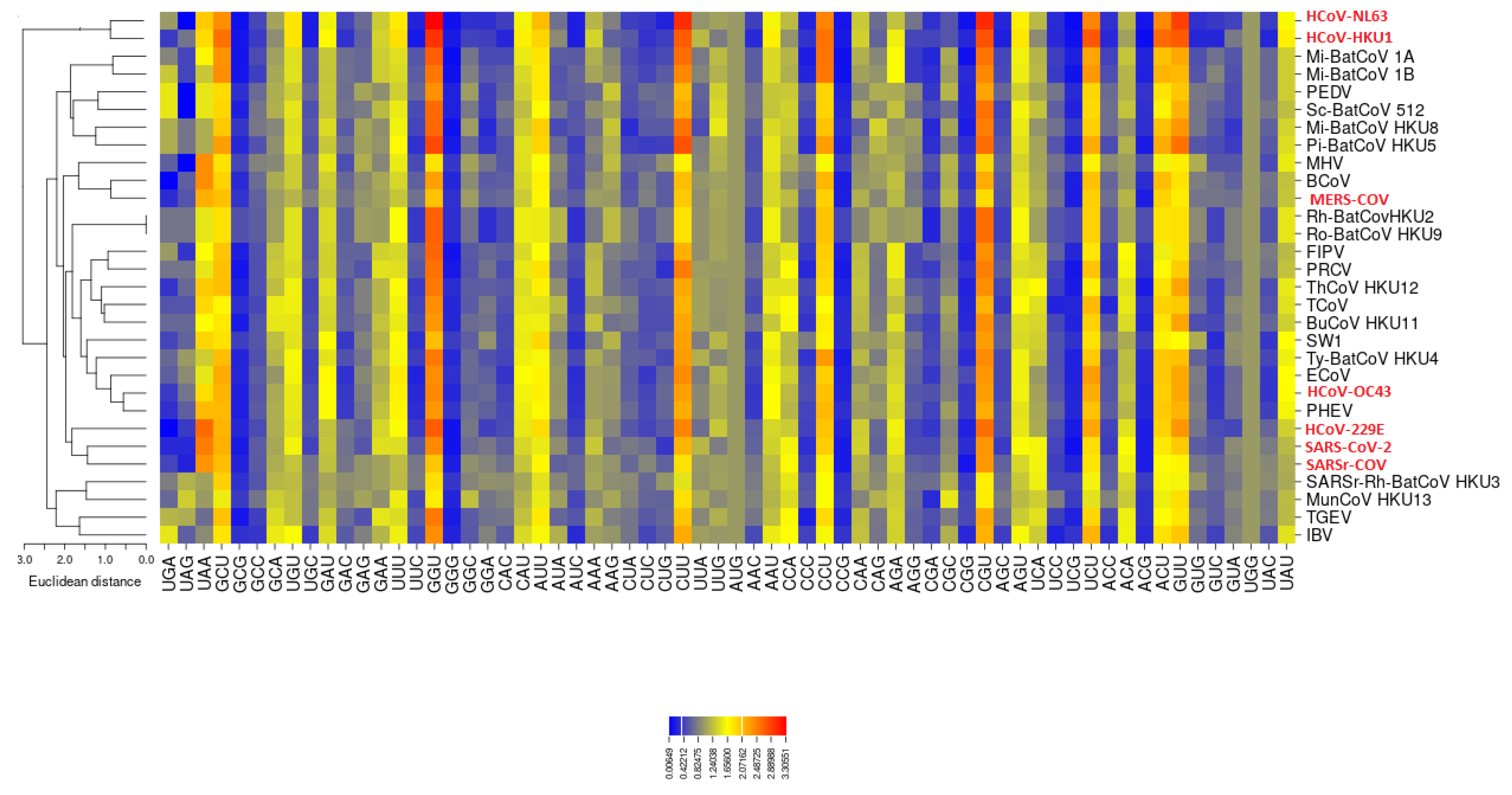
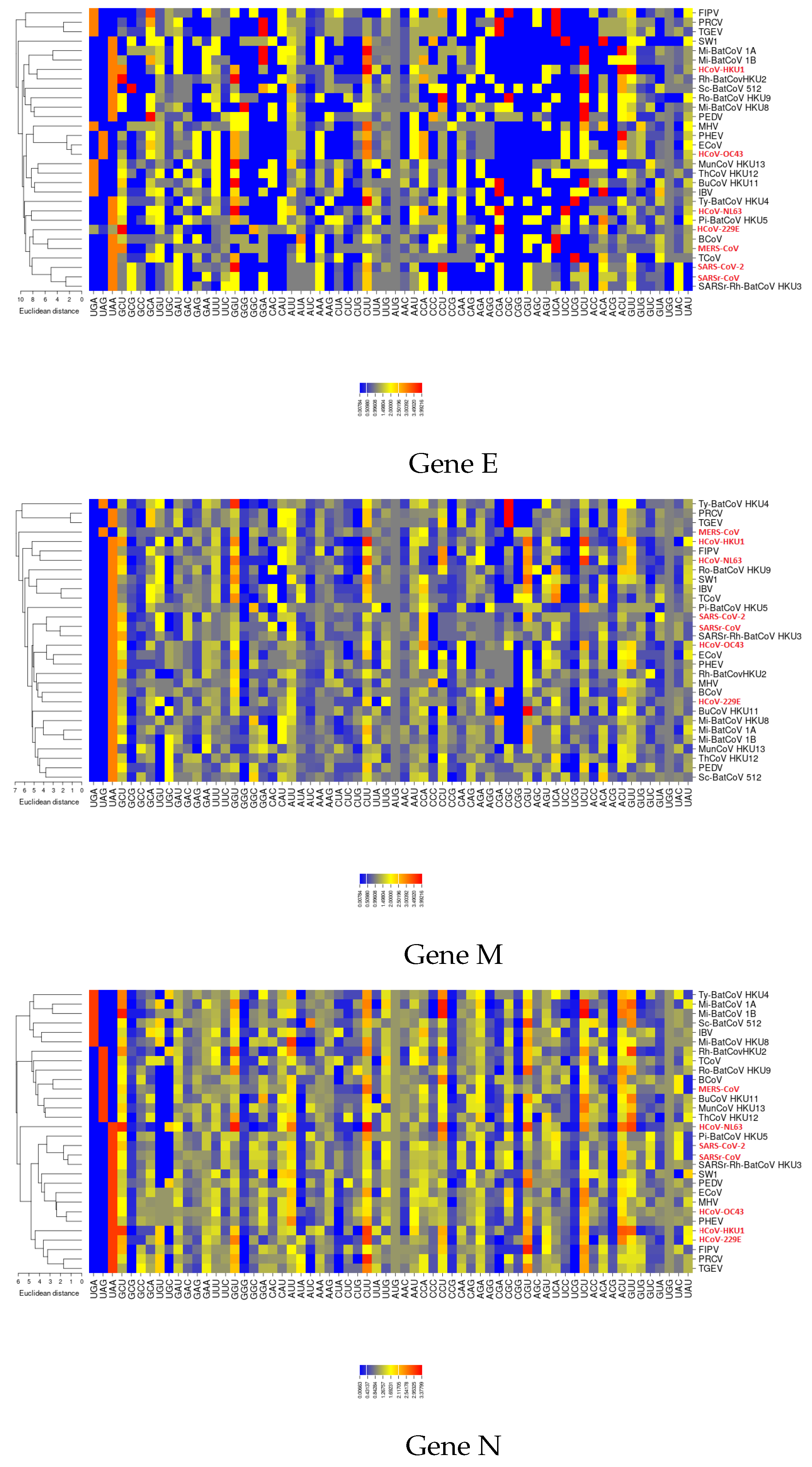
3.2. All the Sequenced SARS-CoV-2 Genomes Share a Common Codon Usage
3.3. Codon Usage of SARS-CoV-2
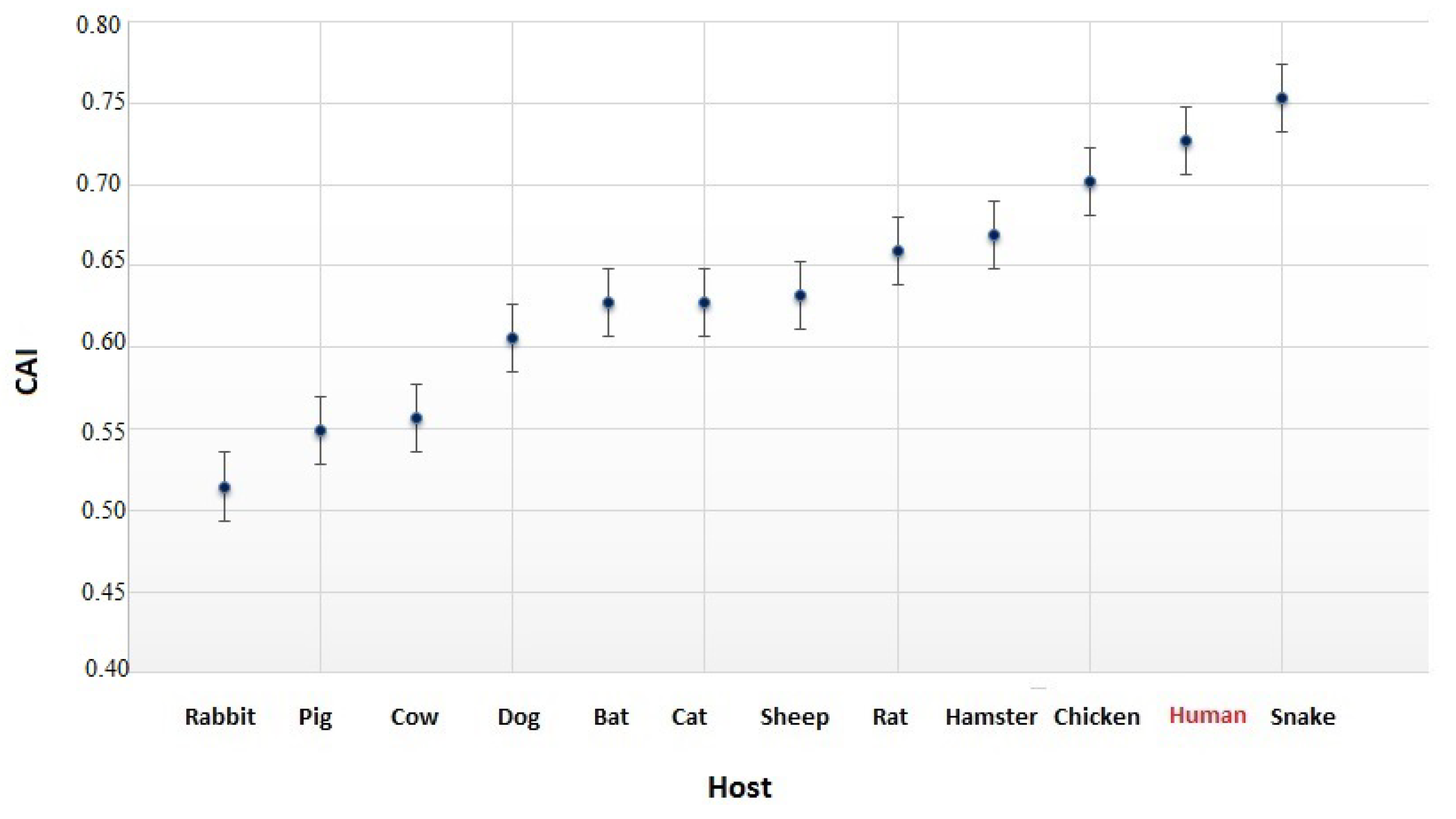
3.4. The Codon Usage of SARS-CoV-2 in Relation to the Human Host
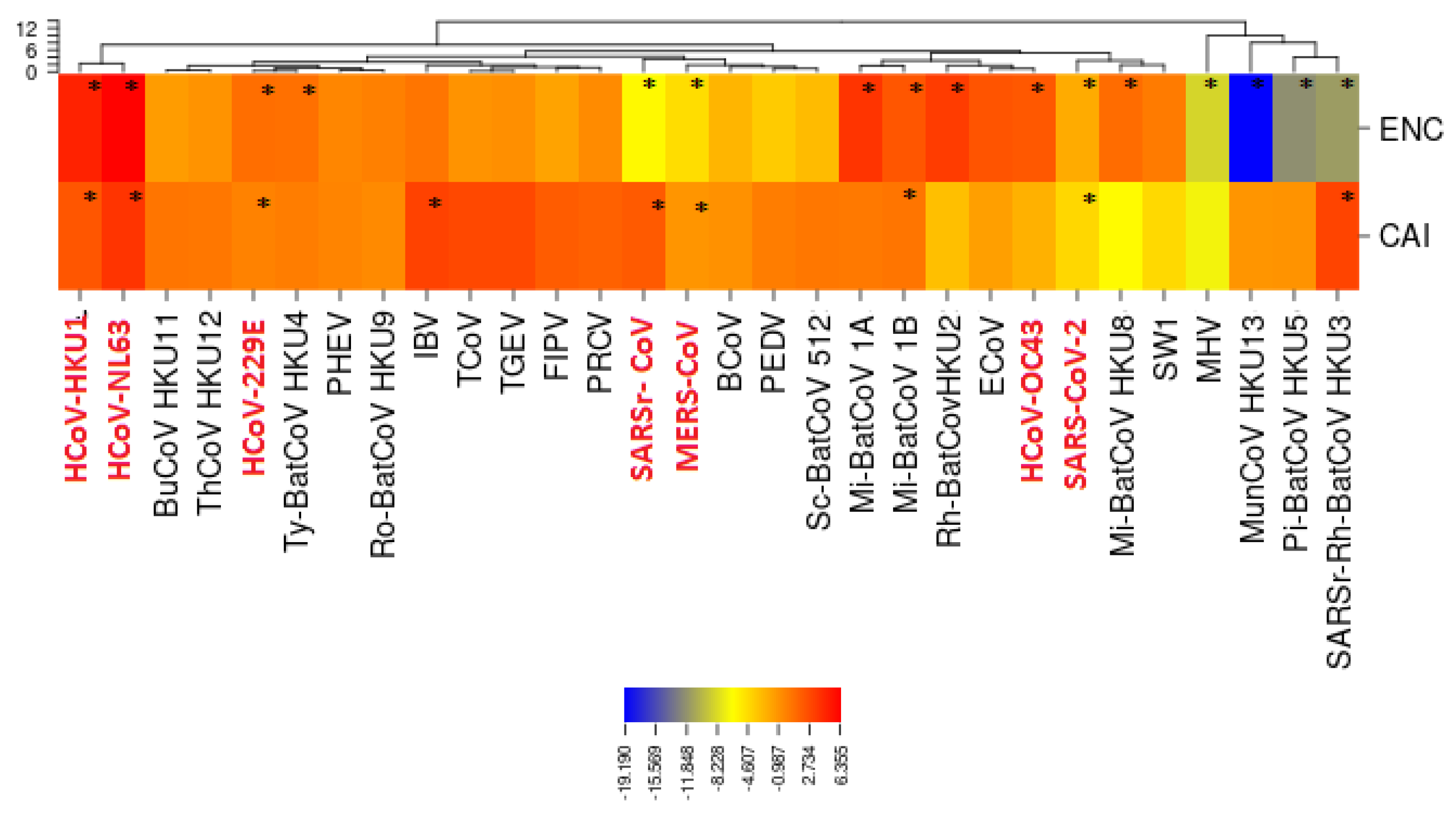
3.5. Selective Pressures and Mutational Rates Characterizing Five Conserved Coronavirus Genes
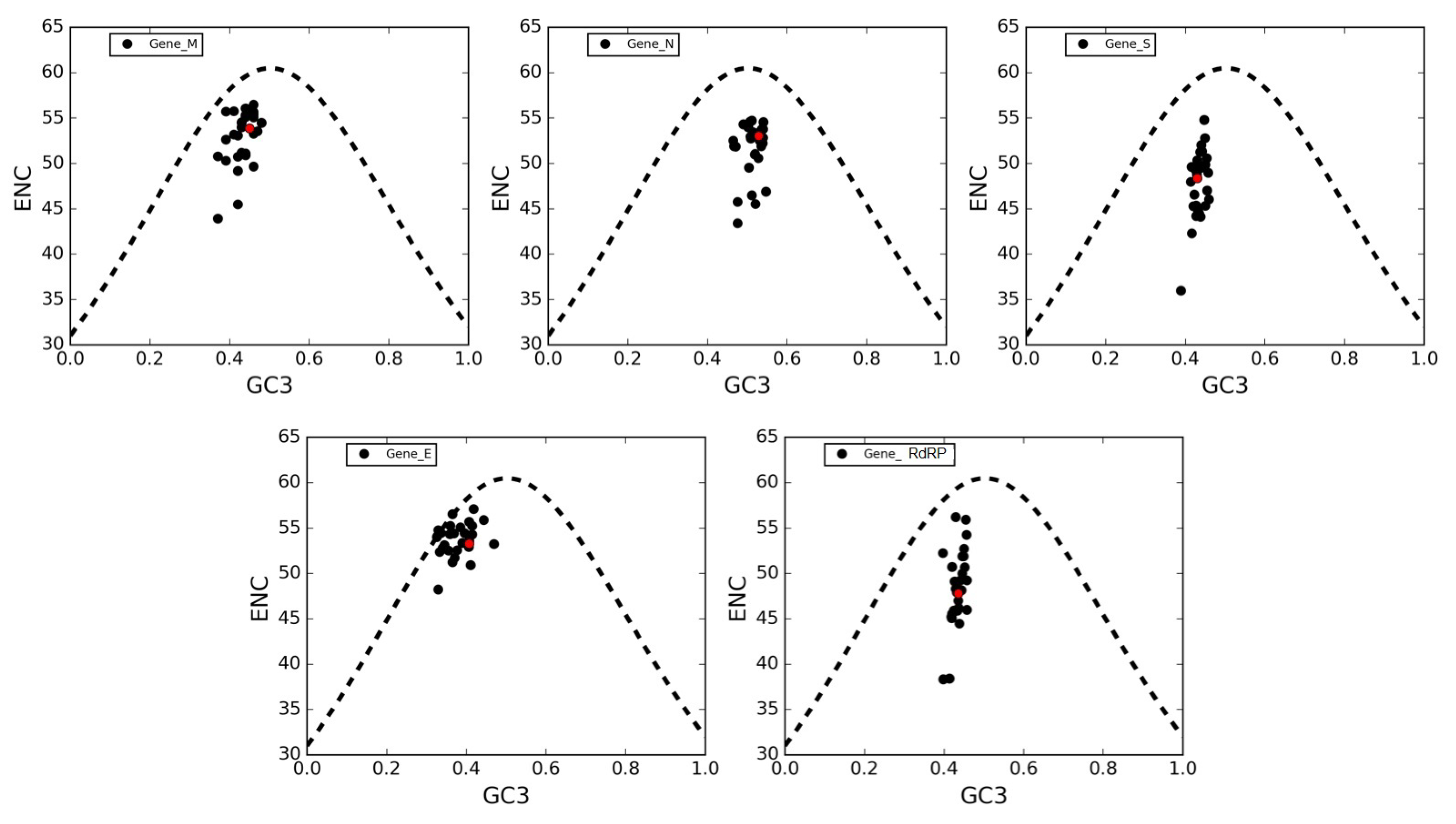
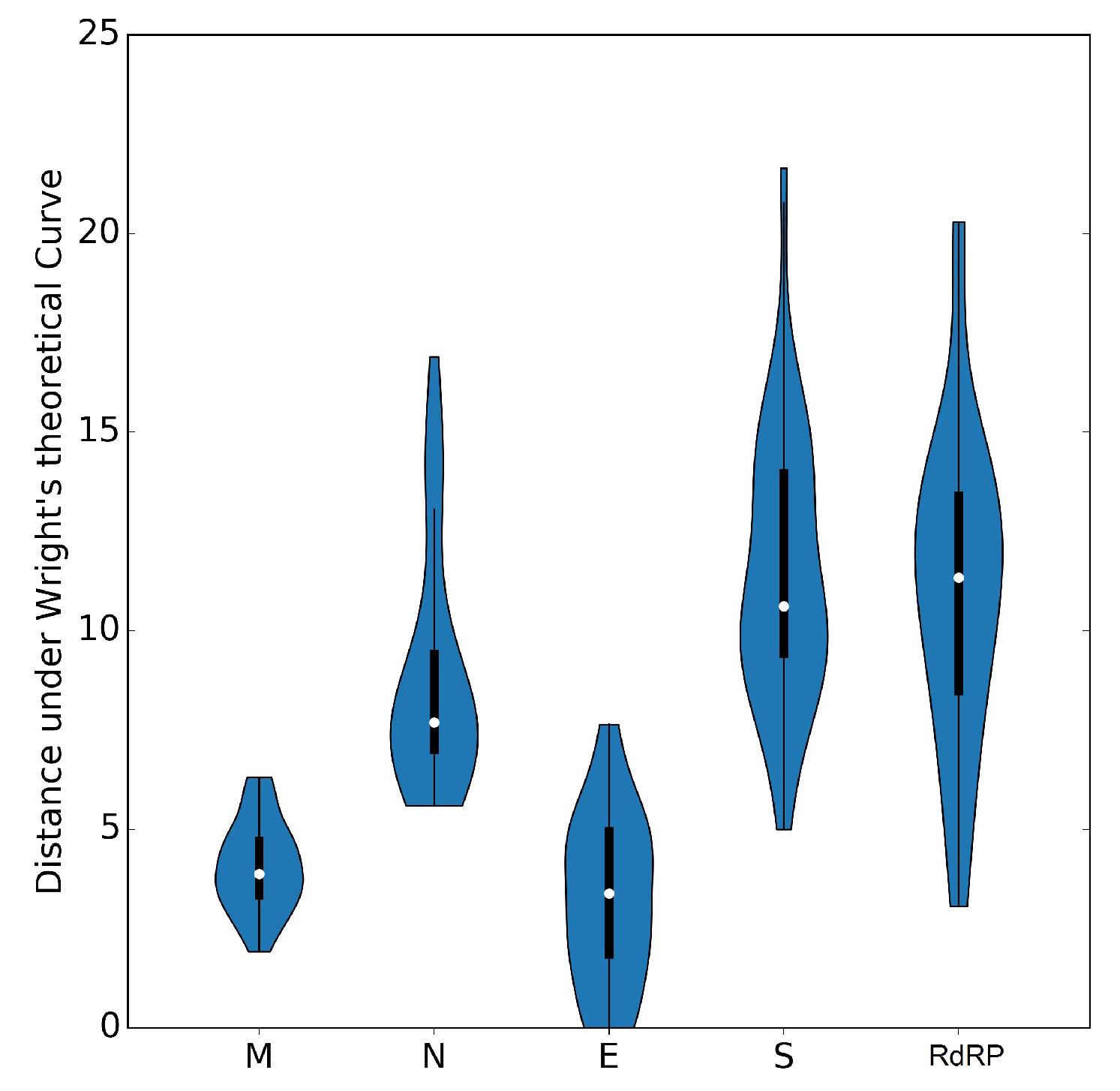
3.6. The ENC Plot Analysis of Individual Genes of SARS-CoV-2
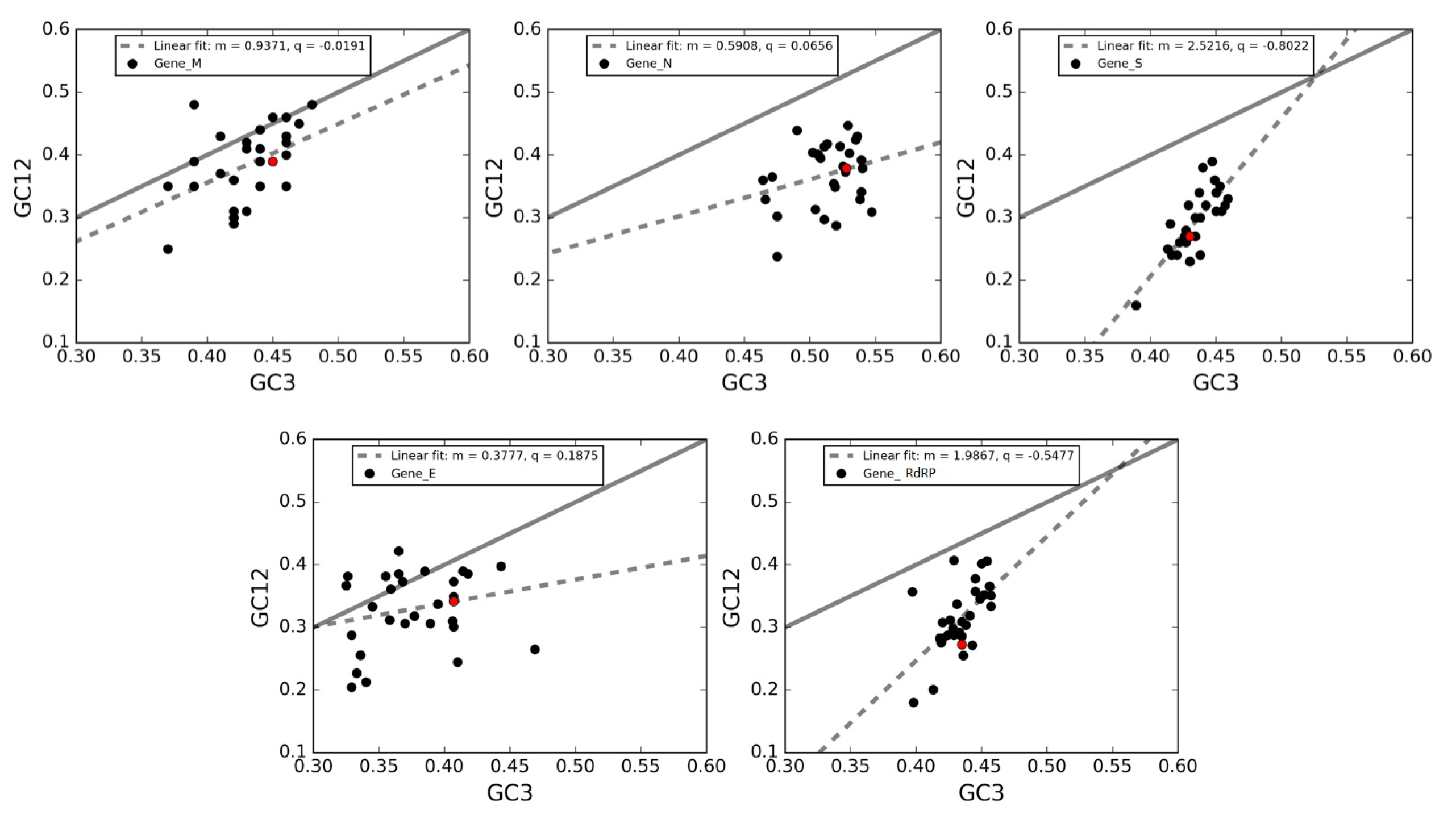
3.7. Neutrality Plot of Individual Genes of SARS-CoV-2
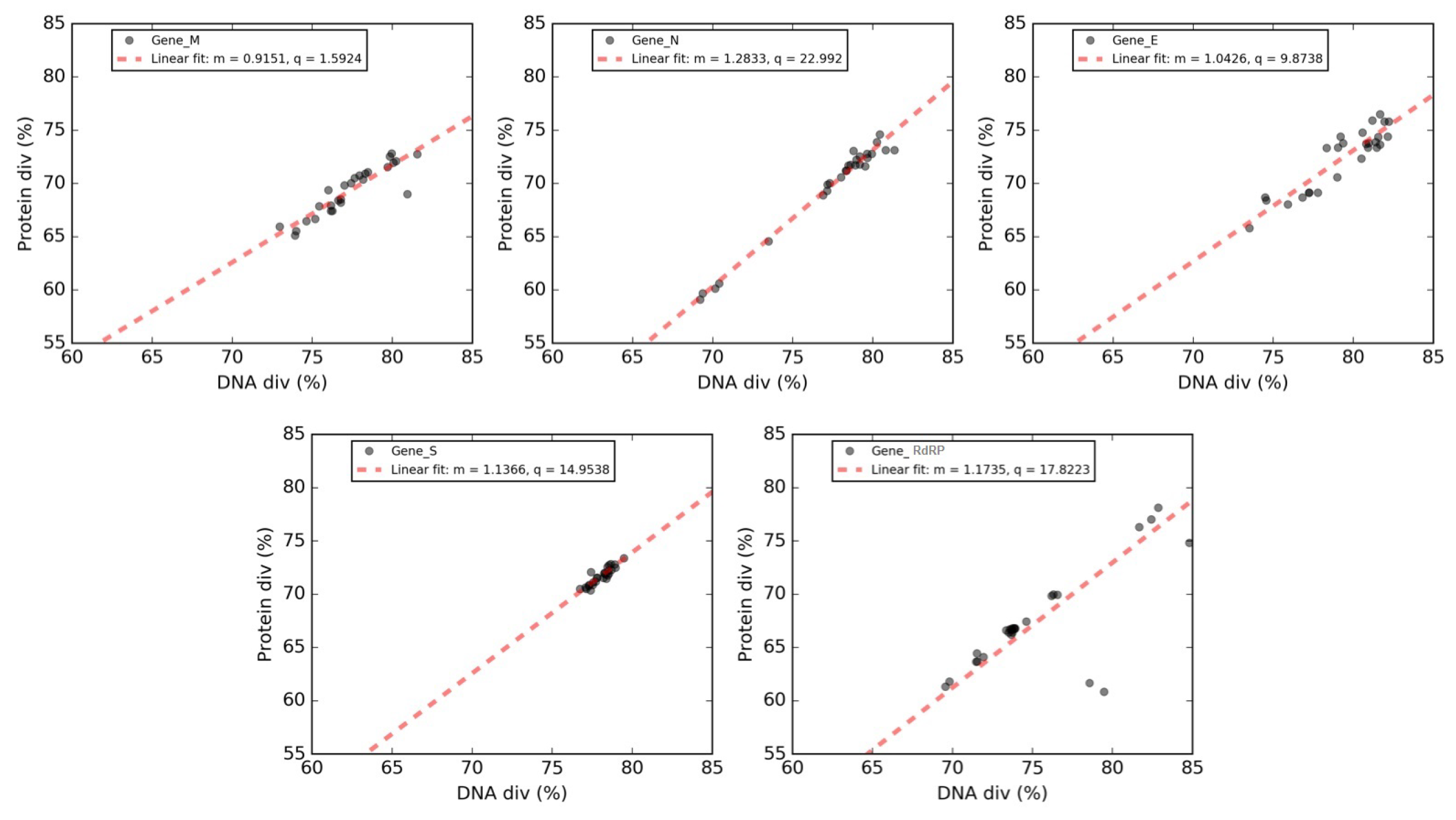
3.8. Forsdyke Plot of Individual Genes of SARS-CoV-2
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A





References
- Lai, M.M.C. Coronavirus: Organization, replication and expression of genome. Annu. Vet. Microbiol. 1990, 44, 303–333. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Siddell, S.G.; Ziebuhr, J.; Snijder, E.J. Coronaviruses, Toroviruses, and Arteriviruses. Topley Wilson’s Microbiol. Microb. Infect. 2005. [Google Scholar] [CrossRef]
- Woo, P.C.; Huang, Y.; Lau, S.K.; Yuen, K.Y. Coronavirus genomics and bioinformatics analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, R.A.; Kuiken, T.; Schutten, M.; Van Amerongen, G.; Van Doornum, G.J.; Van den Hoogen, B.G.; Peiris, M. Aetiology: Koch’s postulates fulfilled for SARS virus. Nature 2003, 423, 240. [Google Scholar] [CrossRef]
- Van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van, B.S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaev, A.A. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; Al-Taher, A.; Al-Nazawi, M.; Al-Mubarak, A.I.; Kandeel, M. Analysis of preferred codon usage in the coronavirus N genes and their implications for genome evolution and vaccine design. J. Virol. Methods 2020, 277, 113806. [Google Scholar] [CrossRef] [PubMed]
- Belalov, I.S.; Lukashev, A.N. Causes and Implications of Codon Usage Bias in RNA Viruses. PLoS ONE 2013, 8, e56642. [Google Scholar] [CrossRef] [PubMed]
- Dilucca, M.; Cimini, G.; Giansanti, A. Essentiality, conservation, evolutionary pressure and codon bias in bacterial genomes. Gene 2018, 663, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Forcelloni, S.; Giansanti, A. Evolutionary Forces and Codon Bias in Different Flavors of Intrinsic Disorder in the Human Proteome. J. Mol. Evol. 2020, 88, 164–178. [Google Scholar] [CrossRef]
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pavé, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, r49–r62. [Google Scholar] [CrossRef]
- Jenkins, G.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Q.; Yuan, X.; Li, X. Analysis of the codon usage pattern in Middle East Respiratory Syndrome Coronavirus. Oncotarget 2017, 8, 110337–110349. [Google Scholar] [CrossRef]
- Sharp, P.M.; Wen-Hsiung, L. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Myers, T.G.; O’Connor, P.M.; Friend, S.H.; Fornace, A.J., Jr.; Kohn, K.W.; Fojo, T.; Bates, S.E.; Rubinstein, L.V.; Anderson, N.L.; et al. An information-intensive approach to the molecular pharmacology of cancer. Science 1997, 275, 343–349. [Google Scholar] [CrossRef]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Wen-Hsiung, L. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Lia, G.; Wang, H.; Wanga, S.; Xinga, G.; Zhanga, C.; Zhanga, W. Insights into the genetic and host adaptability of emerging porcine circovirus. Virulence 2018, 9, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ’effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef]
- Forcelloni, S.; Giansanti, A. Mutations in disordered proteins as early indicators of nucleic acid changes triggering speciation. Sci. Rep. 2020, 10, 4467. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinform. 2014, 48, 3–13. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Berkhout, B.; Van Hemert, F. On the biased nucleotide composition of the human coronavirus RNA genome. Virus Res. 2015, 202, 41–47. [Google Scholar] [CrossRef]
- Woo, P.C.; Wong, B.H.; Huang, Y.; Lau, S.K.; Yuen, K.Y. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology 2007, 369, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wang, W.; Zhao, X.; Zai, J.; Li, X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Callaway, E.; Cyranoski, D. Why snakes probably aren’t spreading the new China virus. Nature 2020, 577, 1. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, W.; Bell, E.W.; Zhou, X.; Zhang, Y. Protein Structure and Sequence Reanalysis of 2019-nCoV Genome Refutes Snakes as Its Intermediate Host and the Unique Similarity between Its Spike Protein Insertions and HIV-1. J. Proteome Res. 2020, 19, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.Y.; Shum, M.H.H.; Zhu, H.C.; Tong, Y.G.; Ni, X.B.; Liao, Y.S.; Wei, W.; Cheung, W.Y.M.; Li, W.J.; Li, L.F.; et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 2020. [Google Scholar] [CrossRef]
- Ceraolo, C.; Giorgi, F.M. Genomic variance of the 2019-nCoV coronavirus. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, Y.; Huang, Y.; Yuen, K.Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Cavanagh, D. The Coronavirus Surface Glycoprotein. In The Coronaviridae; Springer: Boston, MA, USA, 1995; pp. 73–113. [Google Scholar]
- Ruch, T.; Machamer, C. The coronavirus E protein: assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Timani, K.A.; Ye, L.; Ye, L.; Zhu, Y.; Wu, Z.; Gong, Z. Cloning, sequencing, expression, and purification of SARS-associated coronavirus nucleocapsid protein for serodiagnosis of SARS. J. Clin. Virol. 2004, 30, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Ghadimi-Moghadam, A.; Haghani, M.; Bevelacqua, J.J.; Jafarzadeh, A.; Kaveh-Ahangar, A.; Mortazavi, S.M.J.; Ghadimi-Moghadam, A.; Mortazavi, S.A.R. COVID-19 Tragic Pandemic: Concerns over Unintentional “Directed Accelerated Evolution” of Novel Coronavirus (SARS-CoV-2) and Introducing a Modified Treatment Method for ARDS. J. Biomed. Phys. Eng. 2020, 10, 241–246. [Google Scholar]
- SeungHye, H.; Rama, K.M. The acute respiratory distress syndrome: from mechanism to translation. J. Immunol. 2015, 194, 855–860. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The experience of clinical immunologists from China. Clin Immunol. 2020, 108393. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Dhawan, G. How radiotherapy was historically used to treat pneumonia: Could it be useful today? Yale J. Biol. Med. 2013, 86, 555–570. [Google Scholar]
- Kirby, C.; Mackenzie, M. Is low dose radiation herapy a potential treatment for COVID-19 pneumonia? Radiother. Oncol. 2020. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Name | Abbreviation | NCBI Code |
|---|---|---|---|
| alphacoronavirus | Feline infectious peritonitis virus | FIPV | NC_002306.3 |
| alphacoronavirus | Human coronavirus 229E | HCoV-229E | NC_002645.1 |
| alphacoronavirus | Human coronavirus NL63 | HCoV-NL63 | NC_005831.2 |
| alphacoronavirus | Miniopterus bat coronavirus 1A | Mi-BatCoV 1A | NC_010437.1 |
| alphacoronavirus | Miniopterus bat coronavirus 1B | Mi-BatCoV 1B | EU420137.1 |
| alphacoronavirus | Miniopterus bat coronavirus HKU8 | Mi-BatCoV HKU8 | NC_010438.1 |
| alphacoronavirus | Porcine epidemic diarrhea virus | PEDV | NC_003436.1 |
| alphacoronavirus | Porcine respiratory coronavirus | PRCV | DQ811787.1 |
| alphacoronavirus | Rhinolophus bat coronavirus HKU2 | Rh-BatCovHKU2 | NC_009988.1 |
| alphacoronavirus | Scotophilus bat coronavirus 512 | Sc-BatCoV 512 | NC_009657.1 |
| alphacoronavirus | Transmissible gastroenteritis virus | TGEV | NC_038861.1 |
| betacoronavirus | Bovine coronavirus | BCoV | NC_003045.1 |
| betacoronavirus | Equine coronavirus | ECoV | LC061274.1 |
| betacoronavirus | Human coronavirus HKU1 | HCoV-HKU1 | NC_006577.2 |
| betacoronavirus | Human coronavirus OC43 | HCoV-OC43 | NC_006213.1 |
| betacoronavirus | Mouse hepatitis virus | MHV | NC_001846.1 |
| betacoronavirus | Porcine hemagglutinating encephalomyelitis virus | PHEV | DQ011855.1 |
| betacoronavirus | Severe acute respiratory syndrome-related coronavirus 2 | SARS-CoV-2 | NC_045512.2 |
| betacoronavirus | Severe acute respiratory syndrome-related coronavirus | SARSr-CoV | NC_004718.3 |
| betacoronavirus | SARS-related Rhinolophus bat coronavirus HKU3/ | SARSr-Rh-BatCoV HKU3 | NC_009694.1 |
| betacoronavirus | Middle East respiratory syndrome-related coronavirus | MERS-CoV | NC_019843.3 |
| betacoronavirus | Bat coronavirus HKU9-1 | Ro-BatCoV HKU9 | NC_009021.1 |
| betacoronavirus | Pipistrellus bat coronavirus HKU5 | Pi-BatCoV HKU5 | NC_009020.1 |
| betacoronavirus | Tylonycteris bat coronavirus HKU4 | Ty-BatCoV HKU4 | NC_009019.1 |
| gammacoronavirus | Avian infectious bronchitis virus | IBV | NC_001451.1 |
| gammacoronavirus | Beluga whale coronavirus SW1 | SW1 | NC_010646.1 |
| gammacoronavirus | Turkey coronavirus | TCoV | NC_010800.1 |
| deltacoronavirus | Bulbul coronavirus HKU11-934 | BuCoV HKU11 | NC_011547.1 |
| deltacoronavirus | Munia coronavirus HKU13-3514 | MunCoV HKU13 | NC_011550.1 |
| deltacoronavirus | Thrush coronavirus HKU12-600 | ThCoV HKU12 | NC_011549.1 |
| A | C | G | U | |
|---|---|---|---|---|
| ObsN | 12688 | 7693 | 8393 | 13709 |
| Freq. | 0.30 | 0.18 | 0.20 | 0.32 |
| Abbr. | ENC | CAI |
|---|---|---|
| BCoV | 52.10 ± 2.36 | 0.69 ± 0.04 |
| BuCoV HKU11 | 51.41 ± 1.85 | 0.68 ± 0.04 |
| ECoV | 49.31 ± 4.02 | 0.691 ± 0.02 |
| FIPV | 51.56 ± 1.99 | 0.67 ± 0.048 |
| HCoV-HKU1 | 44.58 ± 7.33 | 0.67 ± 0.02 |
| HCoV-229E | 50.29 ± 3.62 | 0.68 ± 0.02 |
| HCoV-NL63 | 44.67 ± 5.35 | 0.66 ± 0.03 |
| HCoV-OC43 | 49.57 ± 3.66 | 0.692 ± 0.02 |
| IBV | 50.65 ± 2.90 | 0.65 ± 0.05 |
| MERS-CoV | 53.08 ± 2.53 | 0.69 ± 0.03 |
| MHV | 53.62 ± 1.72 | 0.71 ± 0.02 |
| Mi-BatCoV 1A | 48.23 ± 3.81 | 0.68 ± 0.03 |
| Mi-BatCoV 1B | 49.31 ± 4.11 | 0.68 ± 0.03 |
| Mi-BatCoV HKU8 | 50.12 ± 4.14 | 0.70 ± 0.02 |
| MunCoV HKU13 | 53.96 ± 0.86 | 0.69 ± 0.04 |
| PEDV | 52.44 ± 2.153 | 0.68 ± 0.04 |
| PHEV | 51.09 ± 3.553 | 0.68 ± 0.02 |
| Pi-BatCoV HKU5 | 53.91 ± 1.36 | 0.70 ± 0.04 |
| PRCV | 51.27 ± 3.15 | 0.67 ± 0.03 |
| Rh-BatCovHKU2 | 48.08 ± 4.49 | 0.70 ± 0.02 |
| Ro-BatCoV HKU9 | 50.91 ± 2.31 | 0.68 ± 0.03 |
| SARSr-CoV | 53.64 ± 2.43 | 0.67 ± 0.04 |
| SARS-CoV-2 | 51.98 ± 2.59 | 0.72 ± 0.05 |
| SARSr-Rh-BatCoV HKU3 | 54.30 ± 1.61 | 0.67 ± 0.03 |
| Sc-BatCoV 512 | 52.38 ± 2.63 | 0.68 ± 0.04 |
| SW1 | 50.86 ± 1.791 | 0.70 ± 0.03 |
| TCoV | 51.34 ± 2.31 | 0.66 ± 0.05 |
| TGEV | 51.39 ± 3.27 | 0.67 ± 0.04 |
| ThCoV HKU12 | 51.43 ± 2.83 | 0.68 ± 0.03 |
| Ty-BatCoV HKU4 | 50.37 ± 3.74 | 0.68 ± 0.03 |
| Gene | Y Intercept | Slope | X Intercept |
|---|---|---|---|
| Matrix (M) | 1.59 | 0.91 | −1.74 |
| Envelope (E) | 9.87 | 1.04 | −9.46 |
| Spike surface (S) | 14.95 | 1.14 | −13.15 |
| RNA replicase (RdRP) | 17.82 | 1.18 | −15.19 |
| Nucleocapsid (N) | 23 | 1.30 | −17.93 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilucca, M.; Forcelloni, S.; Georgakilas, A.G.; Giansanti, A.; Pavlopoulou, A. Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes. Viruses 2020, 12, 498. https://doi.org/10.3390/v12050498
Dilucca M, Forcelloni S, Georgakilas AG, Giansanti A, Pavlopoulou A. Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes. Viruses. 2020; 12(5):498. https://doi.org/10.3390/v12050498
Chicago/Turabian StyleDilucca, Maddalena, Sergio Forcelloni, Alexandros G. Georgakilas, Andrea Giansanti, and Athanasia Pavlopoulou. 2020. "Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes" Viruses 12, no. 5: 498. https://doi.org/10.3390/v12050498
APA StyleDilucca, M., Forcelloni, S., Georgakilas, A. G., Giansanti, A., & Pavlopoulou, A. (2020). Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes. Viruses, 12(5), 498. https://doi.org/10.3390/v12050498