Effect of the Viral Hemorrhagic Septicemia Virus Nonvirion Protein on Translation via PERK-eIF2α Pathway

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. VHSV Infection
2.3. VHSV Amplification and Purification
2.4. Chemicals, Plasmids and Antibodies
2.5. Virus Yield and IFN Bioassays
2.6. Transfection
2.7. Luciferase Assays
2.8. Immunoblotting
2.9. Click-iT OPP Alexa Fluor 488 Imaging and Immunofluorescence Microscopy
2.10. Real-Time Quantitative PCR
2.11. Statistics
3. Results
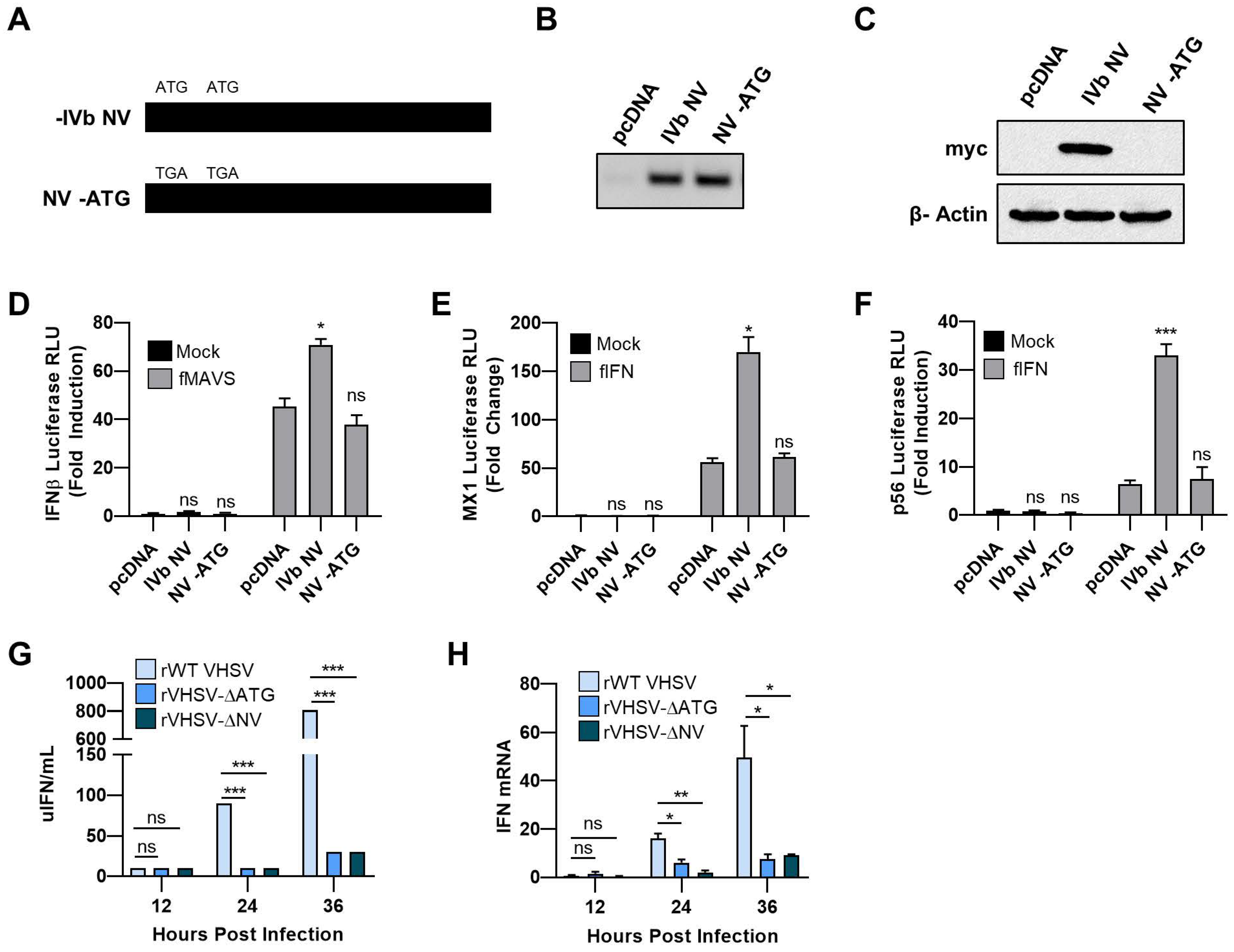
3.1. VHSV IVb NV Protein Augments IFN Signaling
3.2. IVb NV Deficient VHSV Infection Inhibits IFN Signaling
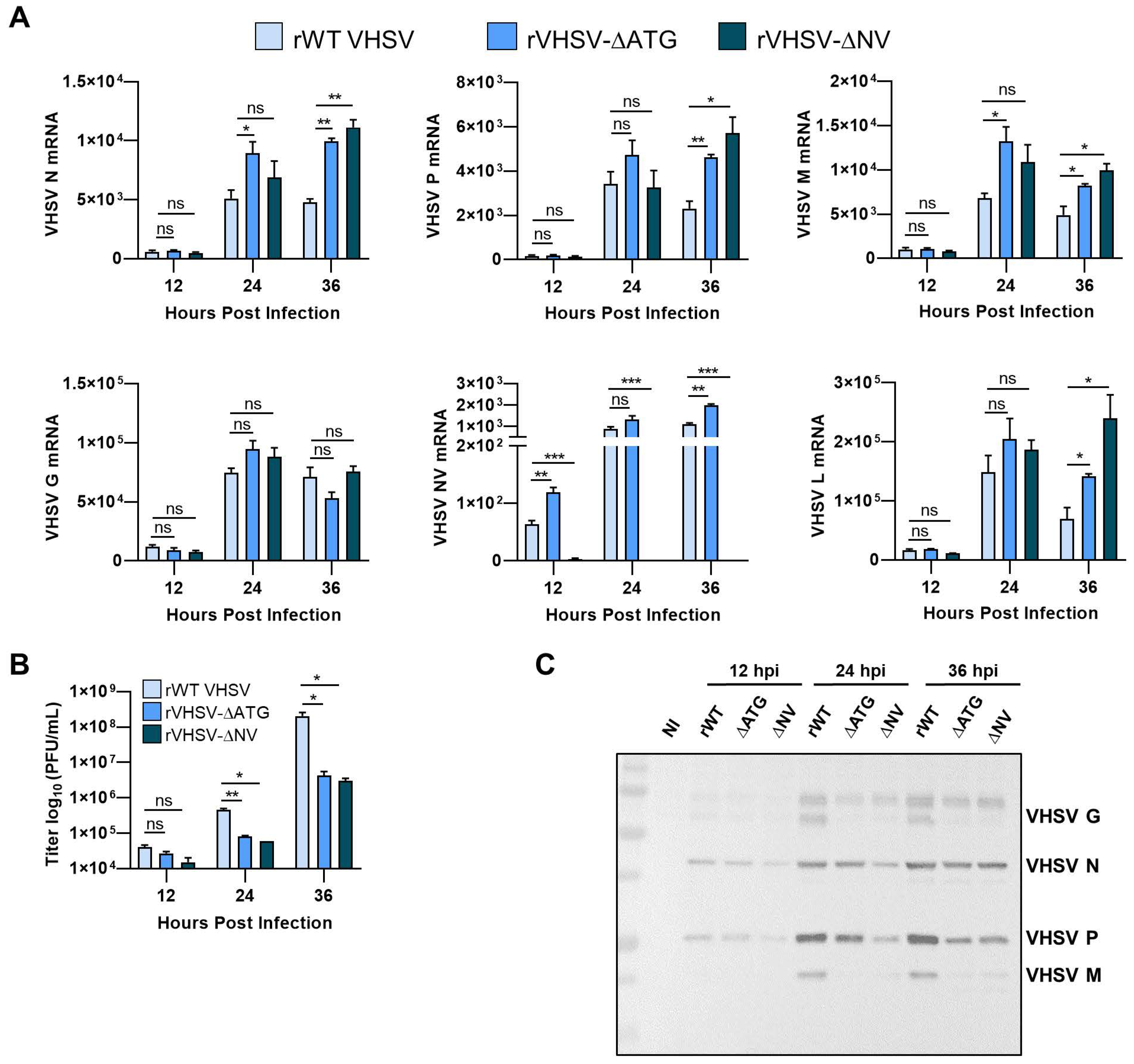
3.3. IVb NV Is Required for Efficient Viral Protein Synthesis
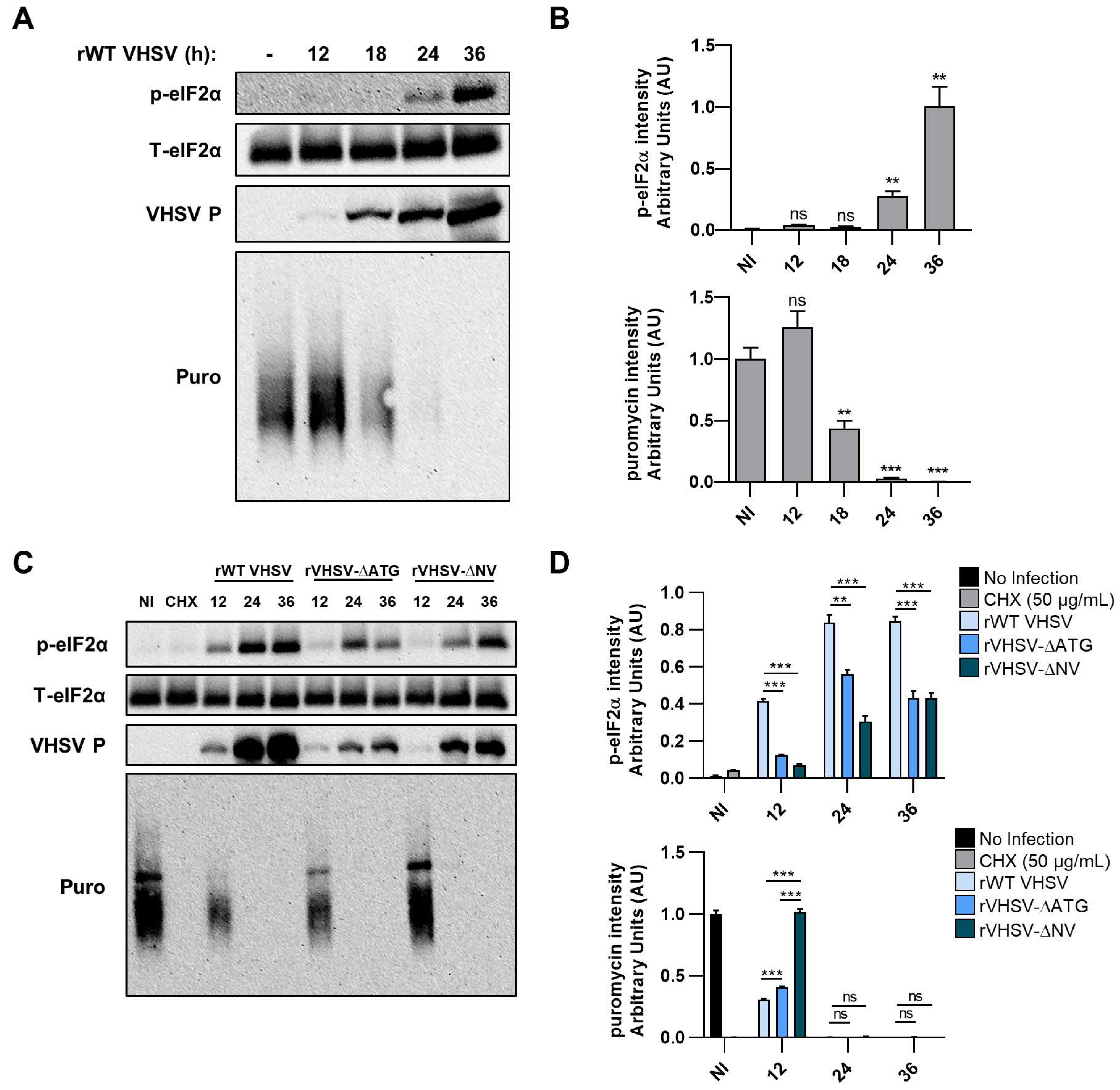
3.4. IVb NV Deficient VHSV Inhibits Translation
3.5. VHSV IVb Infection Inhibits Host Translation and Phosphorylates eIF2α
3.6. Inhibiting PERK Abolishes VHSV IVb-Induced Phosphorylation of eIF2α But Does Not Rescue Host Translation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schutze, H.; Enzmann, P.; Mundt, E.; Mettenleiter, T. Identification of the non-virion (NV) protein of fish rhabdoviruses viral. J. Gen. Virol. 1996, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Einer-Jensen, K.; Ahrens, P.; Forsberg, R.; Lorenzen, N. Evolution of the fish rhabdovirus viral haemorrhagic septicaemia virus. J. Gen. Virol. 2004, 85, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Schutze, H.; Mundt, E.; Mettenleiter, T.C. Complete genomic sequence of viral hemorrhagic septicemia virus, a fish rhabdovirus. Virus Genes 1999, 19, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ammayappan, A.; Vakharia, V.N. Molecular characterization of the Great Lakes viral hemorrhagic septicemia virus (VHSV) isolate from USA. Virol. J. 2009, 6, 171. [Google Scholar] [CrossRef]
- Snow, M.; Cunningham, C.O.; Melvin, W.T.; Kurath, G. Analysis of the nucleoprotein gene identifies distinct lineages of viral haemorrhagic septicaemia virus within the European marine environment. Virus Res. 1999, 63, 35–44. [Google Scholar] [CrossRef]
- Pierce, L.R.; Stepien, C.A. Evolution and biogeography of an emerging quasispecies: Diversity patterns of the fish Viral Hemorrhagic Septicemia virus (VHSv). Mol. Phylogenet. Evol. 2012, 63, 327–341. [Google Scholar] [CrossRef]
- Elsayed, E.; Faisal, M.; Thomas, M.; Whelan, G.; Batts, W.; Winton, J. Isolation of viral haemorrhagic septicaemia virus from muskellunge, Esox masquinongy (Mitchill), in Lake St Clair, Michigan, USA reveals a new sublineage of the North American genotype. J. Fish. Dis. 2006, 29, 611–619. [Google Scholar] [CrossRef]
- Lumsden, J.S.; Morrison, B.; Yason, C.; Russell, S.; Young, K.; Yazdanpanah, A.; Huber, P.; Al-Hussinee, L.; Stone, D.; Way, K. Mortality event in freshwater drum Aplodinotus grunniens from Lake Ontario, Canada, associated with viral haemorrhagic septicemia virus, type IV. Dis. Aquat. Organ. 2007, 76, 99–111. [Google Scholar] [CrossRef]
- Groocock, G.H.; Getchell, R.G.; Wooster, G.A.; Britt, K.L.; Batts, W.N.; Winton, J.R.; Casey, R.N.; Casey, J.W.; Bowser, P.R. Detection of viral hemorrhagic septicemia in round gobies in New York State (USA) waters of Lake Ontario and the St. Lawrence River. Dis. Aquat. Organ. 2007, 76, 187–192. [Google Scholar] [CrossRef]
- Gagne, N.; Mackinnon, A.M.; Boston, L.; Souter, B.; Cook-Versloot, M.; Griffiths, S.; Olivier, G. Isolation of viral haemorrhagic septicaemia virus from mummichog, stickleback, striped bass and brown trout in eastern Canada. J. Fish. Dis. 2007, 30, 213–223. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Mamane, Y.; Hiscott, J. Structural and functional analysis of interferon regulatory factor 3: Localization of the transactivation and autoinhibitory domains. Mol. Cell Biol. 1999, 19, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Stark, G.R. How cells respond to interferons revisited: From early history to current complexity. Cytokine Growth Factor Rev. 2007, 18, 419–423. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Tan, S.L.; Katze, M.G. Translational control of viral gene expression in eukaryotes. Microbiol. Mol. Biol. Rev. 2000, 64, 239–280. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2alpha kinases: Their structures and functions. Cell Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Weaver, W.; Pore, A.; Gorgoglione, B.; Wildschutte, J.H.; Xiao, P.; Shepherd, B.S.; Spear, A.; Malathi, K.; Stepien, C.A.; et al. Role of Viral Hemorrhagic Septicemia Virus Matrix (M) Protein in Suppressing Host Transcription. J. Virol. 2017, 91, e00279-17. [Google Scholar] [CrossRef] [PubMed]
- Biacchesi, S.; Merour, E.; Chevret, D.; Lamoureux, A.; Bernard, J.; Bremont, M. NV Proteins of Fish Novirhabdovirus Recruit Cellular PPM1Bb Protein Phosphatase and Antagonize RIG-I-Mediated IFN Induction. Sci. Rep. 2017, 7, 44025. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, K.H. Effects of NV gene knock-out recombinant viral hemorrhagic septicemia virus (VHSV) on Mx gene expression in Epithelioma papulosum cyprini (EPC) cells and olive flounder (Paralichthys olivaceus). Fish Shellfish Immunol. 2012, 32, 459–463. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, K.H. The role of viral hemorrhagic septicemia virus (VHSV) NV gene in TNF-alpha- and VHSV infection-mediated NF-kappaB activation. Fish Shellfish Immunol. 2013, 34, 1315–1319. [Google Scholar] [CrossRef]
- Chinchilla, B.; Encinas, P.; Estepa, A.; Coll, J.M.; Gomez-Casado, E. Transcriptome analysis of rainbow trout in response to non-virion (NV) protein of viral haemorrhagic septicaemia virus (VHSV). Appl. Microbiol. Biot. 2015, 99, 1827–1843. [Google Scholar] [CrossRef]
- Chinchilla, B.; Encinas, P.; Coll, J.M.; Gomez-Casado, E. Differential Immune Transcriptome and Modulated Signalling Pathways in Rainbow Trout Infected with Viral Haemorrhagic Septicaemia Virus (VHSV) and Its Derivative Non-Virion (NV) Gene Deleted. Vaccines 2020, 8, 58. [Google Scholar] [CrossRef]
- Ammayappan, A.; Kurath, G.; Thompson, T.M.; Vakharia, V.N. A reverse genetics system for the Great Lakes strain of viral hemorrhagic septicemia virus: The NV gene is required for pathogenicity. Mar. Biotechnol. (Ny) 2011, 13, 672–683. [Google Scholar] [CrossRef]
- Ammayappan, A.; Vakharia, V.N. Nonvirion protein of novirhabdovirus suppresses apoptosis at the early stage of virus infection. J. Virol. 2011, 85, 8393–8402. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, D.S.; Kim, K.H. Generation and characterization of NV gene-knockout recombinant viral hemorrhagic septicemia virus (VHSV) genotype IVa. Dis. Aquat. Organ. 2011, 97, 25–35. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yusuff, S.; Kurath, G.; Kim, M.S.; Tesfaye, T.M.; Li, J.; McKenney, D.G.; Vakharia, V.N. The glycoprotein, non-virion protein, and polymerase of viral hemorrhagic septicemia virus are not determinants of host-specific virulence in rainbow trout. Virol. J. 2019, 16, 31. [Google Scholar] [CrossRef]
- Collet, B.; Boudinot, P.; Benmansour, A.; Secombes, C.J. An Mx1 promoter-reporter system to study interferon pathways in rainbow trout. Dev. Comp. Immunol. 2004, 28, 793–801. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Chinchilla, B.; Gomez-Casado, E. Identification of the functional regions of the viral haemorrhagic septicaemia virus (VHSV) NV protein: Variants that improve function. Fish Shellfish Immunol. 2017, 70, 343–350. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, J.A.; Kim, K.H. Effects of a broad-spectrum caspase inhibitor, Z-VAD(OMe)-FMK, on viral hemorrhagic septicemia virus (VHSV) infection-mediated apoptosis and viral replication. Fish Shellfish Immunol. 2016, 51, 41–45. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef]
- Johnson, A.G.; Grosely, R.; Petrov, A.N.; Puglisi, J.D. Dynamics of IRES-mediated translation. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2017, 372, 1716. [Google Scholar] [CrossRef]
- Hanson, P.J.; Zhang, H.M.; Hemida, M.G.; Ye, X.; Qiu, Y.; Yang, D. IRES-Dependent Translational Control during Virus-Induced Endoplasmic Reticulum Stress and Apoptosis. Front. Microbiol. 2012, 3, 92. [Google Scholar] [CrossRef]
- Mauro, V.P.; Edelman, G.M. The ribosome filter redux. Cell Cycle 2007, 6, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Muhs, M.; Yamamoto, H.; Ismer, J.; Takaku, H.; Nashimoto, M.; Uchiumi, T.; Nakashima, N.; Mielke, T.; Hildebrand, P.W.; Nierhaus, K.H.; et al. Structural basis for the binding of IRES RNAs to the head of the ribosomal 40S subunit. Nucleic Acids Res. 2011, 39, 5264–5275. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Burdeinick-Kerr, R.; Whelan, S.P. A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Prangley, E.; Donovan, J.; Demarest, K.; Wingreen, N.S.; Meir, Y.; Korennykh, A. Concerted 2-5A-Mediated mRNA Decay and Transcription Reprogram Protein Synthesis in the dsRNA Response. Mol. Cell 2019, 75, 1218–1228.e6. [Google Scholar] [CrossRef]
- Burke, J.M.; Moon, S.L.; Matheny, T.; Parker, R. RNase L Reprograms Translation by Widespread mRNA Turnover Escaped by Antiviral mRNAs. Mol. Cell 2019, 75, 1203–1217.e5. [Google Scholar] [CrossRef]
- Ivanov, I.; Yabukarski, F.; Ruigrok, R.W.; Jamin, M. Structural insights into the rhabdovirus transcription/replication complex. Virus Res. 2011, 162, 126–137. [Google Scholar] [CrossRef]
- Connor, J.H.; Lyles, D.S. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 2002, 76, 10177–10187. [Google Scholar] [CrossRef]
- Connor, J.H.; Lyles, D.S. Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 2005, 280, 13512–13519. [Google Scholar] [CrossRef]
- Neidermyer, W.J., Jr.; Whelan, S.P.J. Global analysis of polysome-associated mRNA in vesicular stomatitis virus infected cells. PLoS Pathog. 2019, 15, e1007875. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′→3′) | Restriction Site |
|---|---|---|
| VHSV IVbNV-ATG2 se | ACGAATTCTAGACGATCCAGCCGGCACACAGCACAACCAG CTTCTCTCCACTTGTCCTCCACGAGTAGATCGCATAC | EcoR1 |
| VHSV IVbNV as | GGTACCCAGTGAAGGAGACTCAGAGC | Kpn1 |
| Primer Name | Sequence (5′→3′) |
|---|---|
| VHSV IVb N se | ACGGATCCAAAACGCAGATCAG |
| VHSV IVb N as | AGGGGTGAGTATACAGTGGAGT |
| VHSV IVb P qRT as | TGTTGTCGGTCTTCTTCCCG |
| VHSV IVb P qRT se | TGAGGCGTATCAAGCTGTCC |
| VHSV IVb L qRT se | CCAGTGACCCTCCGGATCTA |
| VHSV IVb L qRT as | CGATTCCACCCATCAACCCA |
| VHSV IVb M qRT as | ACTGGGCTTGAGTCCAGGTA |
| VHSV IVb M qRT se | TCAACCCCCTGGTTCACCTA |
| VHSV IVb G qRT as | AGCTTTGCTTCCAGGATGGT |
| VHSV IVb G qRT se | TCCGTTATCAGTCACCAGCG |
| VHSV IVb NV qRT se | CAGCACAACCAGCTTCTCTCC |
| VHSV IVb NV qRT as | GTTGAGGTAGTTGCTTGGGTCA |
| EPC IFN se | TGGGTGGAAAATATCCTGAG |
| EPC IFN as | CTCCTTATGTGATGGCTGGT |
| Fish β-actin se | AGACATCAGGGTGTCATGGTTGGT |
| Fish β-actin as | GGGGTGCTCCTCTGGGGCAA |
| GFP se | ATGGTGAGCAAGGGCGAGGA |
| GFP as | TAGCGGCTGAAGCACTGCACGCC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kesterson, S.P.; Ringiesn, J.; Vakharia, V.N.; Shepherd, B.S.; Leaman, D.W.; Malathi, K. Effect of the Viral Hemorrhagic Septicemia Virus Nonvirion Protein on Translation via PERK-eIF2α Pathway. Viruses 2020, 12, 499. https://doi.org/10.3390/v12050499
Kesterson SP, Ringiesn J, Vakharia VN, Shepherd BS, Leaman DW, Malathi K. Effect of the Viral Hemorrhagic Septicemia Virus Nonvirion Protein on Translation via PERK-eIF2α Pathway. Viruses. 2020; 12(5):499. https://doi.org/10.3390/v12050499
Chicago/Turabian StyleKesterson, Shelby Powell, Jeffery Ringiesn, Vikram N. Vakharia, Brian S. Shepherd, Douglas W. Leaman, and Krishnamurthy Malathi. 2020. "Effect of the Viral Hemorrhagic Septicemia Virus Nonvirion Protein on Translation via PERK-eIF2α Pathway" Viruses 12, no. 5: 499. https://doi.org/10.3390/v12050499
APA StyleKesterson, S. P., Ringiesn, J., Vakharia, V. N., Shepherd, B. S., Leaman, D. W., & Malathi, K. (2020). Effect of the Viral Hemorrhagic Septicemia Virus Nonvirion Protein on Translation via PERK-eIF2α Pathway. Viruses, 12(5), 499. https://doi.org/10.3390/v12050499