Abstract
Dengue is a mosquito-borne disease that is of major importance in public health. Although it has been extensively studied at the molecular level, sequencing of the 5′ and 3′ ends of the untranslated regions (UTR) commonly requires specific approaches for completion and corroboration. The present study aimed to characterize the 5′ and 3′ ends of dengue virus types 1 to 4. The 5′ and 3′ ends of twenty-nine dengue virus isolates from acute infections were amplified through a modified protocol of the rapid amplification cDNA ends approach. For the 5′ end cDNA synthesis, specific anti-sense primers for each serotype were used, followed by polyadenylation of the cDNA using a terminal transferase and subsequent PCR amplification with oligo(dT) and internal specific reverse primer. At the 3′ end of the positive-sense viral RNA, an adenine tail was directly synthetized using an Escherichia coli poly(A) polymerase, allowing subsequent hybridization of the oligo(dT) during cDNA synthesis. The incorporation of the poly(A) tail at the 5′ and 3′ ends of the dengue virus cDNA and RNA, respectively, allowed for successful primer hybridization, PCR amplification and direct sequencing. This approach can be used for completing dengue virus genomes obtained through direct and next-generation sequencing methods.
1. Introduction
Dengue virus (DENV), the etiological agent of dengue fever, has been considered one of the most important vector-borne viruses in the tropical world, where an estimated of 390 million infections occur each year [1, 2,3]. A high proportion (around 75%) of DENV infections are asymptomatic, with the remaining 50–100 million infections ranging from dengue without/with warning signs to severe dengue, a life-threatening condition requiring precise clinical management [2].
DENV is a serocomplex of four closely related serotypes (DENV-1 to -4), belonging to the family Flaviviridae, genus Flavivirus [4,5]. The genome consists of a positive-sense single-stranded RNA of approximately 11 kb, which encodes for three structural (capsid, pre-membrane and envelope) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins. At the 5′ and 3′ untranslated regions (UTR), DENV contains several evolutionarily conserved structural RNA elements involved in viral protein synthesis, genome replication, the regulation of viral and host processes and pathogenesis [6].
The 5′ UTR has a length of 95 to 101 nucleotides and contains two structural domains, the stem-loop A (SLA) domain directly interacts with the viral RNA-dependent RNA polymerase (RdRp) [7], while the stem-loop B (SLB) contains a 16-nucleotide element (5′ Upstream AUG Region-5′ UAR), that together with other RNA elements and structures located in the capsid-coding region (5′ Downstream AUG Region-5′ DAR and 5′ cyclization sequence-5′ CS) are involved in genome cyclization promoting viral RNA synthesis [8,9]. Other RNA elements downstream the AUG region are critical for viral functions; the capsid hairpin (cHP) participates in viral protein translation and RNA synthesis and the conserved capsid-coding region 1 (CCR1) is responsible for virus assembly and viral particle production [10,11].
The 3′ UTR, lacking a poly(A) tail, contains evolutionarily conserved RNA structures and regulatory elements involved in the virus-host relationship. It has been divided into three regions (I–III). Region I contains two stem-loops structures (SLI and SLII) located downstream the stop codon of the viral polyprotein; region II contains two dumbbell structures (DB1 and DB2); and region III contains two RNA structures, sHP and the highly conserved 3′ stem-loop structure (3′ SL), with approximately 79 nucleotides in length at the end of the genome [12]. Several cis-acting elements in the 3′ UTR, including pseudoknots (PKI, PKII, PKIII and PKIV) and sequences complimentary to the 5′ region leading to viral RNA circularization (3′CS, 3′DAR, 3′UAR) have been described [9]. They have important roles in modulating viral replication and host processes [13].
DENV genome has been extensively characterized at the molecular level, which is crucial for comparative analyses attempting to identify genetic determinants of disease severity and dengue pathogenesis. However, complete and corroborated sequencing of the 3′ end of single-stranded positive sense viral RNA genomes lacking a poly(A) tail represents a challenge. Technical approaches of most of the commercially available kits for characterization of the 3′ ends are indicated for poly(A)-tailed messenger RNA through 3′ random amplification of cDNA ends-PCR (RACE-PCR). Some modifications using the enzymatic activity of T4 RNA ligase and poly(A) polymerase have been previously described for viral and bacterial models, respectively [14,15]. Here, we describe the successful implementation of terminal transferase-based 5′ RACE-PCR and poly(A) polymerase-based 3′ RACE-PCR approaches for extension and sequencing of the ends of the DENV genome.
2. Materials and Methods
2.1. Cell Cultures, Viral Isolation and Titration
Human sera from DENV-infected patients were diluted (1/100) in minimal essential medium (MEM) supplemented with 1% HEPES, 1% NaHCO3, 5% Tryptose and 2% fetal bovine serum. Subsequently, C6/36 cell cultures were inoculated with 200-µL of each diluted serum. The adhesion step was carried out at 28 °C for 1 h, then 800 µL of MEM supplemented with 2% fetal bovine serum was added. The cultures were incubated at 28 °C and the supernatants collected when cytopathic effect (CPE) was observed (typically syncytia or cell growth arrest), or 9 days post-infection. A second passage for samples without showing CPE was performed by transferring a 200-µL aliquot of the cell supernatant from the first passage to a fresh C6/36 culture. Culture supernatants were collected, cleared, aliquoted and stored at −70 °C. Virus titer was obtained by plaque assay in BHK-21 (ATCC, CRL-12071 ™) monolayers. For this, 7 serial dilutions 1:10 of the culture supernatant were made in MEM. Next, 200 µL of each dilution was added to BHK-21 cells grown in 24-well dishes, at a density of 2 × 105 cells/well and incubated at 37 °C, 5% CO2 for 1 h. Subsequently, 1.5 mL of 1.5% carboxymethyl-cellulose (CMC) in MEM supplemented with 2% FBS was added and incubated for 8 days at 37 °C in 5% CO2, to allow plaque formation. For plaque counting, the cell monolayers were stained with 0.2% crystal violet solution in 3.5% formaldehyde and 35% ethanol. DENV titer was expressed as plaque-forming units (PFU)/mL.
2.2. Viral RNA Extraction and DENV Serotyping
Viral RNA was extracted from 140 μL of culture supernatants using the QIAamp Viral RNA Mini Kit (Qiagen Inc., Chatsworth, CA, USA), following the manufacturer’s recommendations. Purified RNA was stored at −70 °C for subsequent applications. DENV serotyping was performed through a conventional fourplex RT-PCR, as previously described [16].
2.3. Primer Design for Amplification of the DENV 5′ and 3′ Ends of the DENV Genome
The 5′/3′ RACE kit 2nd generation (Roche Diagnostics GmbH, Mannheim, Germany) was used by following the manufacturer’s recommendations. Briefly, three specific antisense primers (SP1, SP2 and SP3) and two sense primers (SP4 and SP5) were designed for cDNA synthesis and PCR amplification of the 5′ and 3′ ends of all DENV serotypes. Primer design was carried out through the PrimerSelect module of the LaserGene® suite version 8.1 (DNASTAR Inc., Madison, WI, USA.). Genomic sequences GQ868568 (DENV-1), NC_001474 (DENV-2), GU131954 (DENV-3) and GQ868585 (DENV-4) were used as reference. Once the best primer sets were defined (Table 1), homologous sequences of the different serotypes and genotypes of DENV circulating in the Americas were aligned through the ClustalW algorithm implemented in MEGA version 6 [17], in order to assess and consider the genetic variability from each genomic region. Therefore, the design was limited to the circulating genotypes in the Americas, which have been stable through time and geographic distribution [18]. The remaining genotypes were not included, due to the high divergence and limitations for primers degeneracy. Primers were designed by following these criteria: (1) length between 17–24 mer; (2) melting temperature between 48–60 °C; (3) stability of the 3′ pentamer −8,5 Kcal/mol; (4) 7-bp length unique sequence in the 3′ of the primer, (5) free 3′-OH of the primer should be complementary to a second or first codon position (if located in a coding region); (6) inclusion of degenerate sites for those nucleotide positions with representative genetic variability among strains through the region. Degenerated sites were introduced according to the IUPAC nucleotide ambiguity code, available at: [19].

Table 1.
Serotype-specific primers used for amplification and sequencing of the 5′ and 3′ ends of the Dengue virus genome.
2.4. Reverse Transcription and PCR Amplification of the 5′ Region of the DENV Genome
The DENV 5′ end cDNA covering the 5′UTR and part of the coding region was synthesized using the GoScript Reverse Transcription System (GoScript-Promega, Madison, USA), following the manufacturer’s recommendations (Figure 1A). The reaction mixture included between 100–200 ng of total RNA extract, serotype specific anti-sense primer SP1 (0.625 μM), 500 µM dNTPs, 25 U reverse transcriptase, MgCl2 (2.6 mM),1X cDNA synthesis buffer, 5 U RNaseOUT (Life Technologies, Carlsbad, CA, USA) and nuclease-free water, for a final volume of 20 μL. The mixture was incubated for 60 min at 55 °C, then, reverse transcriptase was inactivated by heating at 85 °C for 5 min. The purification of cDNA products was performed using the QIAquick PCR purification kit (Qiagen Inc., Chatsworth, CA, USA) following the manufacturer’s recommendations. Poly (A) tailing of the first-strand cDNA using a recombinant terminal transferase and PCR amplification of dA-tailed cDNA were performed according to the 5′/3′ RACE kit 2nd generation manufacturer’s instructions. The PCR was carried out with the serotype-specific primer SP2 and the oligo d(T) anchor primer (5′-GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTTV3′). The reaction mix contained 5 μL of the polyadenylated cDNA, SP2 (0.25 μM), oligo d(T) anchor primer (0.75 μM), 1.25 U of Taq DNA polymerase (Life Technologies Corp., Carlsbad, CA, USA), dNTPs (0.4 mM), MgCl2 (2 mM), 5 μL of 1× buffer without Mg2+ and nuclease-free water, for a final volume of 50 μL. The thermal profile consisted of a denaturation step at 94 °C for 5 min, followed by 40 cycles of (94 °C for 15 s, primer-specific annealing temperature for 30 s, 72 °C for 30 s), followed by a final extension at 72 °C and 5 m. Expected sizes were 453, 380, 448 and 477 bp for DENV-1 to -4, respectively. Subsequently, a nested PCR was performed using the PCR Anchor primer (5′-GACCACGCGTATCGATGTCGAC-3′) and serotype-specific SP3 reverse primer located upstream of SP2. The reaction mixture included 0.5 μL of the SP2-Oligo d(T) PCR product, SP3 (0.25 μM), anchor primer (0.25 μM), Taq DNA polymerase (1.25 U), dNTPs (400 µM), MgCl2 (2 mM) and 5 mL of 10× buffer, for a final volume of 50 μL. The thermal profile was the same described above. Expected sizes were 310, 300, 298, and 321 bp for DENV-1 to -4, respectively.
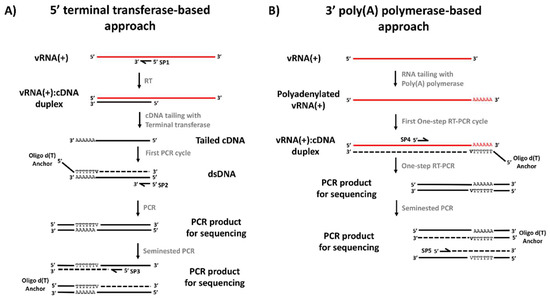
Figure 1.
General strategy for viral genome ends’ amplification and sequencing. (A) Terminal transferase-based protocol for amplification of the 5′ end of the viral genome. (B). Poly(A) polymerase-based protocol for amplification of the 3′ end of the viral genome. V: A, C or G.
2.5. Poly (A) Tailing, Reverse Transcription and PCR Amplification of the End of 3′ Untranslated Region
Given that RNA extracts from infected cell supernatants contain almost exclusively positive-sense DENV genomes and the DENV genome lacks a poly (A) tail, it was not possible to use specific forward primers (SP4 or SP5) or oligo-dT for cDNA synthesis. Therefore, two strategies were defined. The first one consisted in using total RNA obtained from lysates of DENV-infected C6/36 cells (in order to obtain minus strands of the viral RNA) to perform the classic RACE-PCR. The second one was based on the direct A-tailing of positive-sense DENV RNA genomes with 3′-adenine residues, allowing the subsequent annealing of the oligo-dT Anchor Primer (Figure 1B). Briefly, 15 µL of total RNA (~500 ng) were poly-adenylated at the 3′ end in a reaction mixture containing 0.5 U of E. coli poly(A) polymerase system (New England Biolabs, Ipswich, MA, USA), E. coli poly(A) polymerase reaction buffer (50 mM Tris-HCl, 250 mM NaCl, 10 mM MgCl2) and 1mM adenosine-5′-triphosphate (ATP), followed by incubation at 37 °C for 30 min. Afterwards, 5 µL from the reaction were used for a one step RT-PCR using the SuperScript™ III One-Step RT-PCR System (Life Technologies, Carlsbad, CA, USA). Briefly, 50 μL of the reaction were prepared by mixing 25 μL of 2× Reaction Mix, 1 μL of oligo dT-Anchor primer (37.5 μM), 1 μL of specific primer SP4 (12.5 μM), 1 μL of SuperScript™ III RT/Platinum™ Taq Mix, 1 μL of RNaseOUT (5 U/μL) 5 µL of A-tailed RNA and H2O to 50 µL. The thermal profile followed the manufacturer’s recommendations and primer specific annealing temperature. Finally, if no amplification was observed, a nested PCR was performed using the serotype-specific SP5 and the oligo-dT-Anchor primer, with the same conditions described above for the 5′ untranslated region. The expected sizes of the PCR products generated with the SP4 primer were 713, 563, 490 and 452 bp for DENV-1 to -4, respectively; while those for the SP5 primer were 581, 438, 389 and 389 bp for DENV-1 to -4, respectively.
2.6. DNA Sequencing and Sequence Handling
The purification of PCR amplicons was performed using the QIAquick PCR purification kit (Qiagen Inc., Chatsworth, CA, USA), following the manufacturer’s recommendations. For sequencing of the genome ends, reaction mixtures were prepared with 10 ng of each purified PCR product, 2 μL of BigDye Terminator Cycle Sequencing v 3.1 (Applied Biosystems, Carlsbad, CA, USA), 3.2 pmol/μL of the serotype-specific oligonucleotide and 2.0 μL of 5× Sequencing buffer in a final volume of 10 μL. The thermal profile consisted of an initial denaturation at 96 °C for 1 min, followed by 25 cycles of denaturation at 96 °C for 10 s, hybridization at 50 °C for 5 s and extension at 60 °C for 4 min. The products of the sequencing reactions were purified with the BigDye XTerminator purification kit (Applied Biosystems, Carlsbad, CA, USA) and sequenced on the ABI3130 Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA). The sequences were edited and assembled into the SeqMan module of the LaserGene v8.1 suite (DNASTAR Inc. Madison, WI, USA).
3. Results
3.1. Successful Amplification of the 5′ and 3′ Regions of All DENV Serotypes
The 5′ end of Colombian strains corresponding to the four DENV serotypes was successfully amplified by using the standard RACE-PCR approach, that involved the polyadenylation of the cDNA generated with the primer SP1 and subsequent PCR amplification. DENV-2 and -3 were amplified after the first round of PCR amplification with serotype-specific primers SP2, while bands of the expected size were observed for all DENV serotypes after the second round of PCR amplification with serotype-specific primers SP3 (Supplementary Figure S1).
The amplification of the DENV 3′ end was attempted by a similar RACE-PCR strategy, using total RNA extracts from DENV-infected C6/36 cells and forward serotype-specific SP4 primers, which were expected to hybridize the minus strand viral RNA for the cDNA synthesis. Subsequent PCR was unsuccessful, probably due to the asymmetric replication of the DENV genome, and therefore the low abundance of negative-sense viral RNA strands [20]. A second approach based on the poly-adenylation of positive-sense viral RNA by using the E. coli poly (A) polymerase, and subsequent oligo-dT hybridization on the artificially generated poly (A) tail, demonstrated to be successful for the specific amplification of the 3′ ends of all DENV serotypes (Supplementary Figure S2).
3.2. Molecular Characterization of the 5′ and 3′ Ends of the Dengue Virus Genome
The DENV 5′ and 3′ ends of all four DENV serotypes were successfully obtained after Sanger sequencing with specific primers (SP2, SP3, SP4 and SP5). A total of 29 isolates from Colombian clinical samples were successfully sequenced, 26 isolates belonging to patients with dengue without warning signs (3, 10, 10 and 3 from DENV-1 to -4, respectively) and 5 from severe dengue patients (all from DENV-2) (Table 2). Viral titers ranged from 1 × 104 to 2 × 107 PFU/mL. The electropherograms allowed one to confirm the presence of the artificially generated poly (A) tail (Figure 2). At the 3′ end, the last nucleotide is called “N” to designate a variable position; this is a consequence of using a degenerate oligo-dT anchor primer containing A, C or G at the 3′ end. At the 5′ end, the “N” is also present at the -1 position, therefore, it does not affect the sequence of interest.
Table 2.
List of DENV-1, -2, -3 and -4 strains included in the study and associated clinical characteristics.
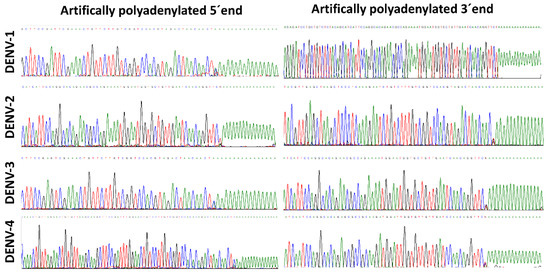
Figure 2.
Artificial polyadenylation at the 5′ and 3′ ends of the dengue virus genome. Electropherograms of the 5′and 3′ ends of representative strains of DENV-1 to DENV-4, obtained through Sanger sequencing. The 5′ and 3′ ends were sequenced by using sequence-specific reverse and forward primers, respectively.
3.3. Complete Coverage at the 5′ and 3′ Ends of the DENV Genome
In order to assess the coverage at the 5′ and 3′ ends, previously reported sequences of DENV strains circulating in South American countries were aligned with those of the present study. A high proportion of DENV “complete genome” records available at GenBank have incomplete or uncorroborated 5′ and 3′ ends, commonly lacking the first or last ~20 nt at the 5′ and 3′ UTR, respectively, due to limitations imposed by current RNA genome amplification techniques by reverse transcription and PCR, which involves the use of primers with previously known sequences during cDNA synthesis, PCR amplification or sequencing. These ends were completely covered by using the strategy described here (Figure 3). Using the BLAST alignment tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi), the sequences obtained in the present study matched with other DENV sequences reported in Latin American and Caribbean countries (Cuba, Haiti, Brazil, Peru, Venezuela, Ecuador, French Guiana, Argentina, Chile and Paraguay).

Figure 3.
DENV 5′ and 3′ ends sequence coverage. (A) Alignment of DENV 5′ end sequences obtained in the present study, with some records of complete genomes available at GenBank. (B) Alignment of DENV 3′ end sequences obtained in the present study, with some records of complete genomes available at GenBank. Black arrowheads denote sequences obtained in the present study.
4. Discussion
During the last two decades, the intense research in fundamental biology of DENV and other closely related flaviviruses has allowed one to decipher the role or function of almost all viral proteins and RNA structural elements during the virus life cycle [11]. These studies have also allowed the identification of several genetic differences that explain some of the phenotypic differences at the virus replication or virus-host interaction and pathogenesis levels, including nucleotide substitutions and insertion/deletions in the coding and the untranslated regions [21,22]. The 5′ and 3′ UTR harbor critical elements for viral RNA replication and translation becoming putative candidates to virulence determinants [23,24,25,26]; for this reason, complete sequencing, including the genome ends, is mandatory for identifying the viral genetic contribution to virulence and severe dengue pathogenesis.
Viral RNA polymerases are error-prone and lack proofreading activity, leading RNA viruses to exhibit extremely high mutation rates [27]. The unavoidable accumulation of genetic variation in the whole DENV genome suppose a challenge for sequence-based molecular methods and therefore justify the rational design of degenerated oligonucleotides that theoretically cover the existent genetic variability and perform well for a longer period of mutation accumulation. Here, we used the PrimerSelect module of the LaserGene suite, which integrates the nearest neighbor algorithm for ΔG and melting temperature (Tm, temperature at which half of the oligonucleotides have hybridized with the sequence) calculation. Using this algorithm, it was possible to design primers that worked well for amplification and sequencing the 5′ and 3′ ends of the DENV genome. All primers were analyzed through sequence alignment to cover the existing variability of DENV strains across the region and to avoid the use of critical primer positions according to evolutionary behavior (e.g., codon position in coding regions).
Some authors have described the complete DENV genome sequencing, despite imposing the primer sequence at the end of the 5′ and 3′ ends [28,29,30]. Other authors have recognized the limitation of the genome ends characterization and have limited their published genomes to actually characterized nucleotides, excluding the primer sequences [31].
The most rigorous approach is well-known as RACE-PCR. The original RACE protocol for the 3′ end was intended to work directly with polyadenylated RNA (e.g., messenger RNA), however, flavivirus genomes lack a poly(A) tail. A first approach to overcome this limitation consisted of applying the RACE strategy to total RNA extracts obtained from cell lysates, in which the presence of negative-sense viral RNA within dsRNA and replicative intermediates was expected. However, the asymmetric replication of the DENV genome and the low abundance of negative-sense viral RNA strands [20] impeded the success of this strategy. Therefore, an approach for the 3′ end characterization was implemented by using the E. coli poly (A) polymerase, which was based on the direct polyadenylation of the positive-sense viral RNA genomes previously extracted from viral particles present in culture supernatants of infected cells. Despite the fact that cellular mRNA transcripts could be present in the extract after viral RNA extraction, the use of virus-specific forward primer after the cDNA synthesis for PCR amplification prevents any interference. The artificially generated poly (A) tail provided a target for the oligo-dT hybridization during the cDNA synthesis and subsequent PCR amplification, following the standard RACE protocol. High quality electropherograms evidenced the successful incorporation of a poly (A) tract to the viral genome and the complete resolution of the 3′ end sequence of the analyzed DENV strains from all four serotypes and from a wide range of virus concentrations.
Novel protocols for the Oxford Nanopore sequencing of RNA virus genomes in their native form have been recently described [32,33]. They employed different approaches for obtaining the starting material and share the artificial polyadenylation step to extend the 3′ end of the viral genome before sequencing. These methods have not been validated in extreme cases of very low RNA concentration or RNA fragmentation/degradation, where a previous amplification step has been shown to improve the performance. Next generation sequencing (NGS) protocols for non-polyadenylated RNA viruses involving amplification require previous knowledge of the sequence for primer hybridization. Therefore, we are extending the method as an easy and useful strategy for genome ends characterization, where PCR amplification is required.
These technical approaches could be implemented in basic molecular biology laboratories and routinely used as a complement for full-genome sequencing of DENV and the general approach may be useful for molecular characterization of the genome ends of whatever virus species with a linear RNA genome. This strategy is compatible with Sanger sequencing or NGS, increasing the coverage at the genome ends and allowing more accurate in comparative genomics studies.
5. Conclusions
The methodological approach was successful for the amplification and sequencing of the 5′ and 3′ ends of the DENV-1 to -4 genomes. The application of this strategy will allow the evaluation of the effect of point mutations on the disruption/conservation of critical RNA structural elements and their potential impact on viral replication and pathogenesis.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4915/12/5/496/s1, Figure S1: PCR amplification of the 5′end of DENV-1 to -4, Figure S2: PCR amplification of the 3′end of DENV-1 to -4.
Author Contributions
Conceptualization, J.A.U.-C.; Investigation, A.R.-M., D.A.A.-D., K.L.-D., D.P.-C.; Methodology, A.R.-M., D.A.A.-D., K.L.-D.; Formal analysis, A.R.-M., D.A.A.-D., K.L.-D., J.A.U.-C.; Writing—original draft, A.R.-M., D.A.A.-D., K.L.-D.; Funding acquisition, J.A.U.-C., K.L.-D.; Supervision, J.A.U.-C.; Writing—review and editing, J.A.U.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by COLCIENCIAS (grant 210465740977 to J.A.U.-C.) and the Colombian National Institute of Health.
Acknowledgments
To the National Laboratory Network for routine virologic surveillance of dengue in Colombia. To the Virology Lab at the Instituto Nacional de Salud, Colombia, for providing the previously diagnosed and serotyped samples, and the Sequencing and Genomics Unit team for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brathwaite Dick, O.; San Martín, J.L.; Montoya, R.H.; del Diego, J.; Zambrano, B.; Dayan, G.H. The History of Dengue Outbreaks in the Americas. Am. J. Trop. Med. Hyg. 2012, 87, 584–593. [Google Scholar] [CrossRef]
- WHO. Dengue Guidelines for Diagnosis, Treatment, Prevention and Control; World Health Organization, Ed.; WHO: Geneva, Switzerland, 2009; pp. 1–160. [Google Scholar]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Twiddy, S.S.; Farrar, J.J.; Vinh Chau, N.; Wills, B.; Gould, E.A.; Gritsun, T.; Lloyd, G.; Holmes, E.C. Phylogenetic relationships and differential selection pressures among genotypes of dengue-2 virus. Virology 2002, 298, 63–72. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.J.; Rice, C.M. Flaviviridae. In Fields in Virology, 6th ed.; Knipe, D.M., Ed.; Williams & Wilkins Lippincott: Philadelphia, PA, USA, 2013; pp. 712–746. [Google Scholar]
- Clyde, K.; Kyle, J.L.; Harris, E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J. Virol. 2006, 80, 11418–11431. [Google Scholar] [CrossRef]
- Filomatori, C.V.; Lodeiro, M.F.; Alvarez, D.E.; Samsa, M.M.; Pietrasanta, L.; Gamarnik, A.V. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 2006, 20, 2238–2249. [Google Scholar] [CrossRef]
- Khromykh, A.A.; Meka, H.; Guyatt, K.J.; Westaway, E.G. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 2001, 75, 6719–6728. [Google Scholar] [CrossRef]
- Friebe, P.; Harris, E. Interplay of RNA elements in the dengue virus 5′ and 3′ ends required for viral RNA replication. J. Virol. 2010, 84, 6103–6118. [Google Scholar] [CrossRef]
- Clyde, K.; Barrera, J.; Harris, E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology 2008, 379, 314–323. [Google Scholar] [CrossRef]
- Gebhard, L.G.; Filomatori, C.V.; Gamarnik, A.V. Functional RNA Elements in the Dengue Virus Genome. Viruses 2011, 3, 1739–1756. [Google Scholar] [CrossRef]
- Villordo, S.M.; Carballeda, J.M.; Filomatori, C.V.; Gamarnik, A.V. RNA Structure Duplications and Flavivirus Host Adaptation. Trends Microbiol. 2016, 24, 270–283. [Google Scholar] [CrossRef]
- Paranjape, S.M.; Harris, E. Y box-binding protein-1 binds to the dengue virus 3′-untranslated region and mediates antiviral effects. J. Biol. Chem. 2007, 282, 30497–30508. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, M.; Zhang, H.; Wang, H.Y.; Wang, L.F. Improved rapid amplification of cDNA ends (RACE) for mapping both the 5′ and 3′ terminal sequences of paramyxovirus genomes. J. Virol. Methods 2005, 130, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Miller, E. Rapid Amplification of cDNA Ends for RNA Transcript Sequencing in Staphylococcus. Methods Mol. Biol. 2016, 1373, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Usme, J.; Gómez, A.; Gallego, J. Molecular detection and typing of dengue virus by RT-PCR and nested PCR using degenerated oligonucleotides. Rev. Salud Uninorte 2012, 28, 1–15. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Usme-Ciro, J.A.; Mendez, J.A.; Laiton, K.D.; Paez, A. The relevance of dengue virus genotypes surveillance at country level before vaccine approval. Hum. Vaccines Immunother. 2014, 10, 2674–2678. [Google Scholar] [CrossRef]
- IUPAC Codes. Available online: https://www.bioinformatics.org/sms2/iupac.html (accessed on 2 April 2020).
- Chu, P.W.; Westaway, E.G. Replication strategy of Kunjin virus: Evidence for recycling role of replicative form RNA as template in semiconservative and asymmetric replication. Virology 1985, 140, 68–79. [Google Scholar] [CrossRef]
- Leitmeyer, K.C.; Vaughn, D.W.; Watts, D.M.; Salas, R.; Villalobos, I.; de Ramos, C.; Rico-Hesse, R. Dengue virus structural differences that correlate with pathogenesis. J. Virol. 1999, 73, 4738–4747. [Google Scholar] [CrossRef]
- Goo, L.; VanBlargan, L.A.; Dowd, K.A.; Diamond, M.S.; Pierson, T.C. A single mutation in the envelope protein modulates flavivirus antigenicity, stability, and pathogenesis. PLoS Pathog. 2017, 13, e1006178. [Google Scholar] [CrossRef]
- Filomatori, C.V.; Carballeda, J.M.; Villordo, S.M.; Aguirre, S.; Pallarés, H.M.; Maestre, A.M.; Sánchez-Vargas, I.; Blair, C.D.; Fabri, C.; Morales, M.A.; et al. Dengue virus genomic variation associated with mosquito adaptation defines the pattern of viral non-coding RNAs and fitness in human cells. PLoS Pathog. 2017, 13, e1006265. [Google Scholar] [CrossRef]
- Sirigulpanit, W.; Kinney, R.M.; Leardkamolkarn, V. Substitution or deletion mutations between nt 54 and 70 in the 5′ non-coding region of dengue type 2 virus produce variable effects on virus viability. J. Gen. Virol. 2007, 88, 1748–1752. [Google Scholar] [CrossRef]
- Tajima, S.; Nukui, Y.; Takasaki, T.; Kurane, I. Characterization of the variable region in the 3′ non-translated region of dengue type 1 virus. J. Gen. Virol. 2007, 88, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.E.; De Lella Ezcurra, A.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.H.; Yip, J.T.; Chen, Y.L.; Liu, W.; Harun, S.; Lystiyaningsih, E.; Heriyanto, B.; Beckett, C.G.; Mitchell, W.P.; Hibberd, M.L.; et al. Periodic re-emergence of endemic strains with strong epidemic potential-a proposed explanation for the 2004 Indonesian dengue epidemic. Infect. Genet. Evol. 2008, 8, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Sasmono, R.T.; Wahid, I.; Trimarsanto, H.; Yohan, B.; Wahyuni, S.; Hertanto, M.; Yusuf, I.; Mubin, H.; Ganda, I.J.; Latief, R.; et al. Genomic analysis and growth characteristic of dengue viruses from Makassar, Indonesia. Infect. Genet. Evol. 2015, 32, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Christenbury, J.G.; Aw, P.P.; Ong, S.H.; Schreiber, M.J.; Chow, A.; Gubler, D.J.; Vasudevan, S.G.; Ooi, E.E.; Hibberd, M.L. A method for full genome sequencing of all four serotypes of the dengue virus. J. Virol. Methods 2010, 169, 202–206. [Google Scholar] [CrossRef]
- Dash, P.K.; Sharma, S.; Soni, M.; Agarwal, A.; Sahni, A.K.; Parida, M. Complete genome sequencing and evolutionary phylogeography analysis of Indian isolates of Dengue virus type 1. Virus Res. 2015, 195, 124–134. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Jenjaroenpun, P.; Taylor, M.K.; Lee, J.; Tolardo, A.L.; Parvathareddy, J.; Kandel, S.; Wadley, T.D.; Kaewnapan, B.; Athipanyasilp, N.; et al. Rapid Sequencing of Multiple RNA Viruses in Their Native Form. Front. Microbiol. 2019, 10, 260. [Google Scholar] [CrossRef]
- Tan, C.C.S.; Maurer-Stroh, S.; Wan, Y.; Sessions, O.M.; de Sessions, P.F. A novel method for the capture-based purification of whole viral native RNA genomes. AMB Express 2019, 9, 45. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).