Phylodynamics Helps to Evaluate the Impact of an HIV Prevention Intervention

 , ,
, , 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Phylogenetic Analysis
2.3. Phylodynamics and Estimation of Epidemiological Parameters
3. Results
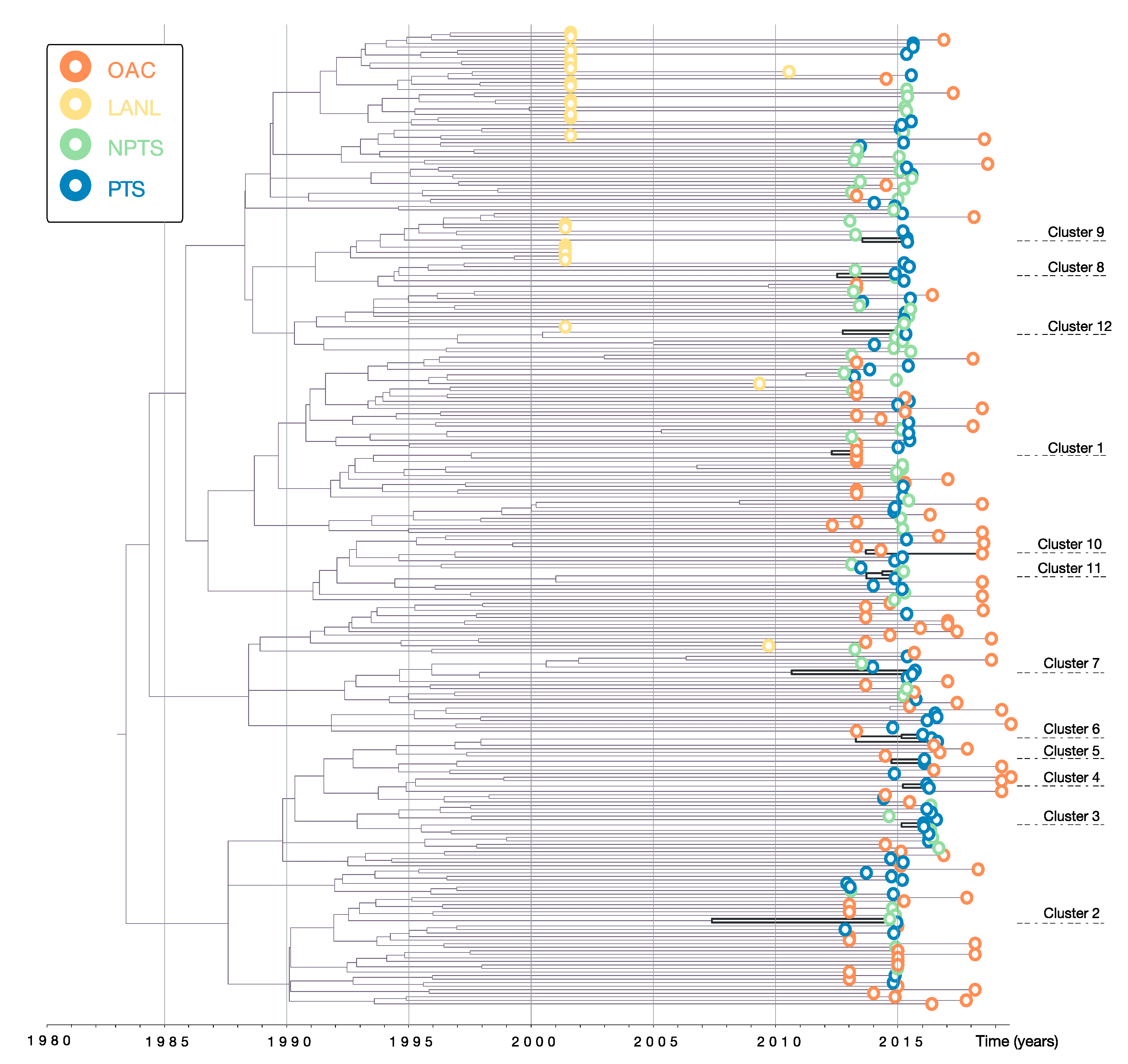
3.1. Data and Phylogenetic Clustering
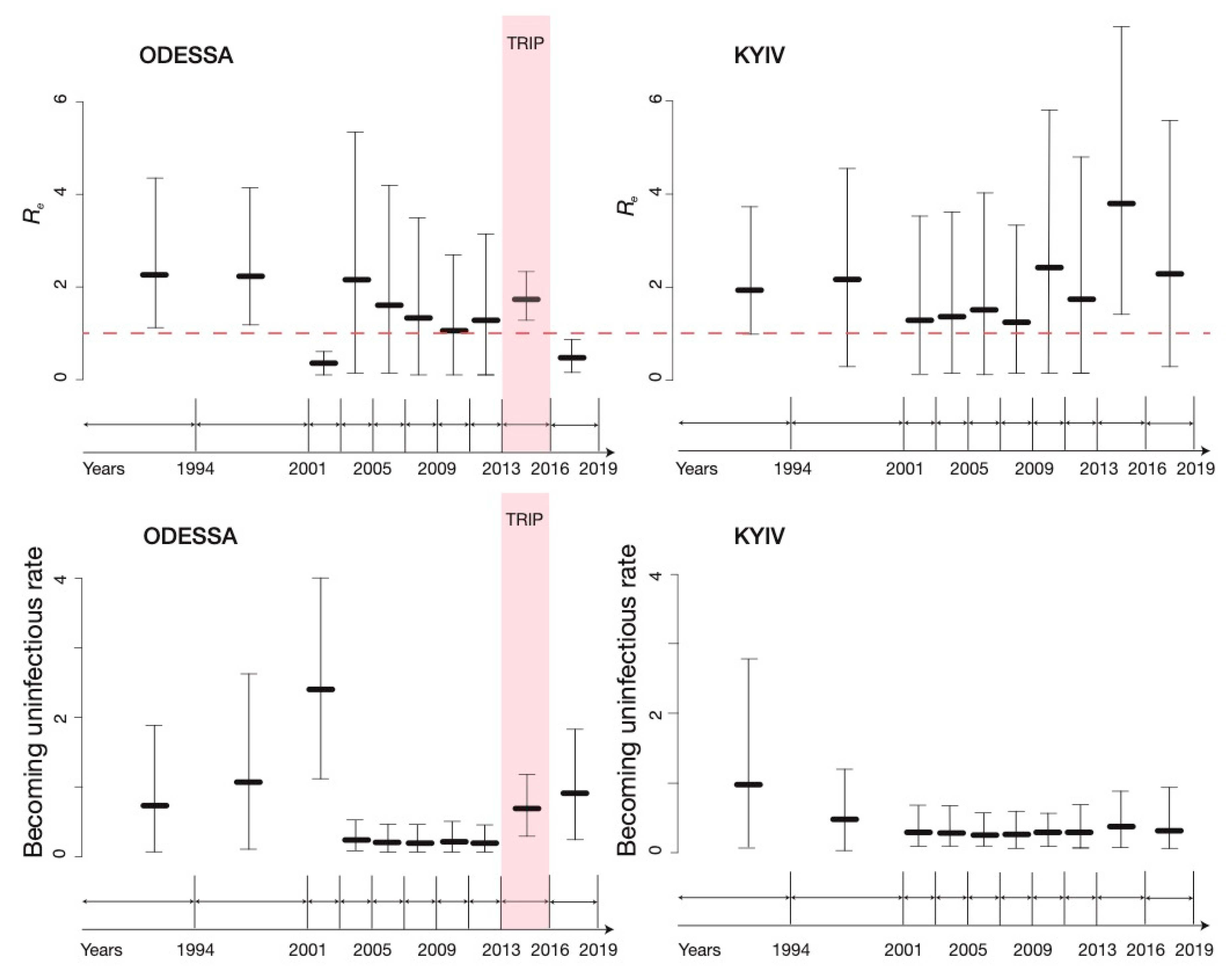
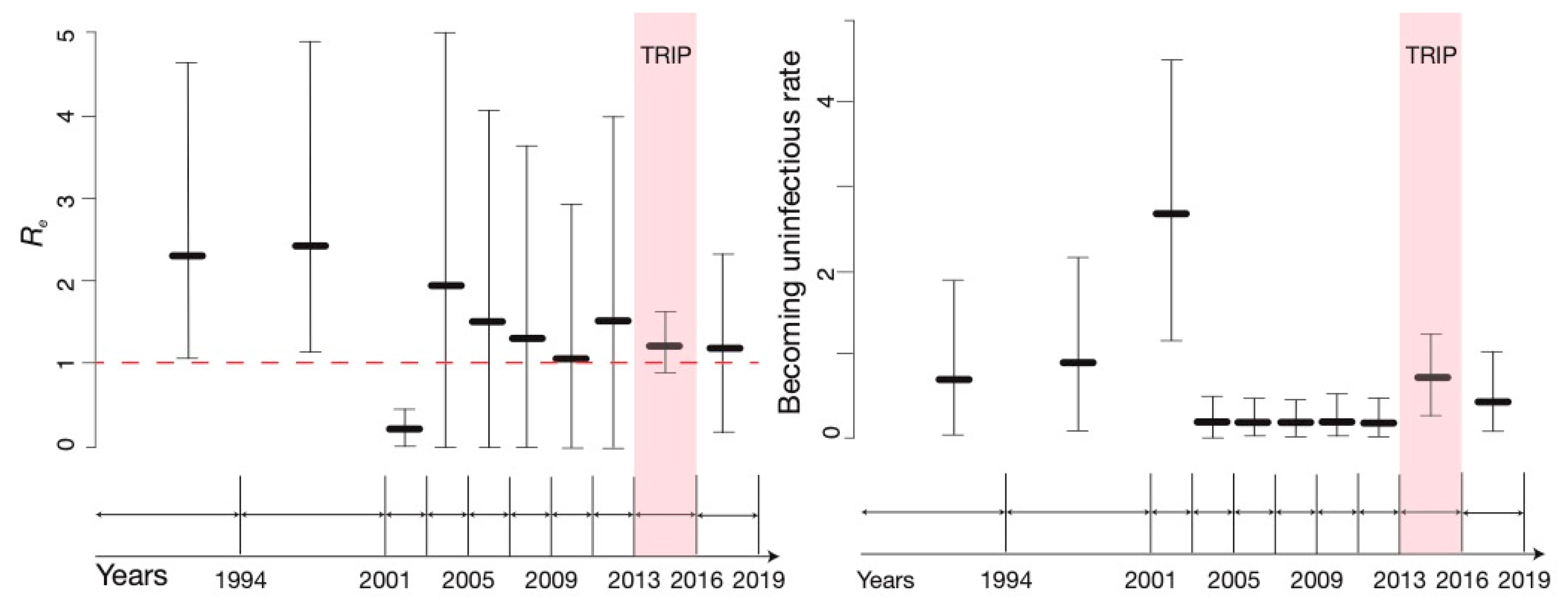
3.2. BDSKY Analysis
3.3. Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Volz, E.M.; Koelle, K.; Bedford, T. Viral phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.F.; Gustafson, R.; Daly, P.; Zerr, L.; Demlow, S.E.; Wong, J.; Woods, C.K.; Hogg, R.S.; Krajden, M.; Moore, D.; et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: An implementation case study. Lancet HIV 2016, 3, e231–e238. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Murrell, B.; Mehta, S.R.; Forgione, L.A.; Kosakovsky Pond, S.L.; Smith, D.M.; Torian, L.V. Growth of HIV-1 Molecular Transmission Clusters in New York City. J. Infect. Dis. 2018, 218, 1943–1953. [Google Scholar] [CrossRef]
- Vasylyeva, T.I.; du Plessis, L.; Pineda-Pena, A.C.; Kuhnert, D.; Lemey, P.; Vandamme, A.M.; Gomes, P.; Camacho, R.J.; Pybus, O.G.; Abecasis, A.B.; et al. Tracing the Impact of Public Health Interventions on HIV-1 Transmission in Portugal Using Molecular Epidemiology. J. Infect. Dis. 2019, 220, 233–243. [Google Scholar] [CrossRef]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pepin, J.; et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Bbosa, N.; Ssemwanga, D.; Nsubuga, R.N.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Nanyonjo, M.; Kuteesa, M.; Seeley, J.; Kiwanuka, N.; Bagaya, B.S.; et al. Phylogeography of HIV-1 suggests that Ugandan fishing communities are a sink for, not a source of, virus from general populations. Sci. Rep.-UK 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Stadler, T.; Kouyos, R.; von Wyl, V.; Yerly, S.; Boni, J.; Burgisser, P.; Klimkait, T.; Joos, B.; Rieder, P.; Xie, D.; et al. Estimating the basic reproductive number from viral sequence data. Mol. Biol. Evol. 2012, 29, 347–357. [Google Scholar] [CrossRef]
- Stadler, T.; Kuhnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl. Acad. Sci. USA 2013, 110, 228–233. [Google Scholar] [CrossRef]
- Kuhnert, D.; Stadler, T.; Vaughan, T.G.; Drummond, A.J. Simultaneous reconstruction of evolutionary history and epidemiological dynamics from viral sequences with the birth-death SIR model. J. R. Soc. Interface 2014, 11, 20131106. [Google Scholar] [CrossRef]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.; et al. Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 2011, 365, 493–505. [Google Scholar] [CrossRef]
- Latkin, C.A.; Davey-Rothwell, M.A.; Knowlton, A.R.; Alexander, K.A.; Williams, C.T.; Boodram, B. Social network approaches to recruitment, HIV prevention, medical care, and medication adherence. J. Acquir. Immune Defic. Syndr. 2013, 63, S54–S58. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.R.; Downing, M.J., Jr.; Smyrnov, P.; Nikolopoulos, G.; Schneider, J.A.; Livak, B.; Magiorkinis, G.; Slobodianyk, L.; Vasylyeva, T.I.; Paraskevis, D.; et al. Socially-integrated transdisciplinary HIV prevention. AIDS Behav. 2014, 18, 1821–1834. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, G.K.; Pavlitina, E.; Muth, S.Q.; Schneider, J.; Psichogiou, M.; Williams, L.D.; Paraskevis, D.; Sypsa, V.; Magiorkinis, G.; Smyrnov, P.; et al. A network intervention that locates and intervenes with recently HIV-infected persons: The Transmission Reduction Intervention Project (TRIP). Sci. Rep. 2016, 6, 38100. [Google Scholar] [CrossRef] [PubMed]
- Smyrnov, P.; Williams, L.D.; Korobchuk, A.; Sazonova, Y.; Nikolopoulos, G.K.; Skaathun, B.; Morgan, E.; Schneider, J.; Vasylyeva, T.I.; Friedman, S.R. Risk network approaches to locating undiagnosed HIV cases in Odessa, Ukraine. J. Int. AIDS Soc. 2018, 21. [Google Scholar] [CrossRef]
- Korobchuk, A.; Davtyan, H.; Denisiuk, O.; Zachariah, R.; Nikolopoulos, G.K.; Paraskevis, D.; Skaathun, B.; Schneider, J.; Vasylyeva, T.I.; Williams, L.D.; et al. People with high HIV viral load within risk networks: Who are these people and who refers them best? J. Infect. Dev. Ctries 2019, 13, 103S–110S. [Google Scholar] [CrossRef]
- Kovtun, O.; Lysenko, Y.; Sokolenko, O.; Mikhedko, A.; kurdus, O.; Korotych, O.; Kuzin, I. The medical information system “HIV-infection in Ukraine”: Development and implementation. Public Health Panor. 2019, 5, 1–121. [Google Scholar]
- Saad, M.D.; Shcherbinskaya, A.M.; Nadai, Y.; Kruglov, Y.V.; Antonenko, S.V.; Lyullchuk, M.G.; Kravchenko, O.N.; Earhart, K.C.; Sanchez, J.L.; Birx, D.L.; et al. Molecular epidemiology of HIV Type 1 in Ukraine: Birthplace of an epidemic. AIDS Res. Hum. Retrovir. 2006, 22, 709–714. [Google Scholar] [CrossRef]
- Barska, Y.G.; Yermolenko, N.Y.; Novak, H.Y.; Sazonova, Y.O.; Strembitska, A.S.; Tonkonog, L.M. Monitoring Behavior and HIV Prevalence among Injecting Drug Users. Regional Profiles Based on the Results of the 2015 Bio-Behavioral Study; Alliance for Public Health: Kyiv, Ukraine, 2016. [Google Scholar]
- Sazonova, Y.; Dukach, Y. Report on the Results of a Bio-Behavioral Study of Men who have Sex with Men in Ukraine; Alliance for Public Health: Kyiv, Ukraine, 2019. [Google Scholar]
- Vasylyeva, T.I.; Friedman, S.R.; Smyrnov, P.; Bondarenko, K. A new approach to prevent HIV transmission: Project Protect intervention for recently infected individuals. AIDS Care 2015, 27, 223–228. [Google Scholar] [CrossRef]
- Vasylyeva, T.I.; Liulchuk, M.; Friedman, S.R.; Sazonova, I.; Faria, N.R.; Katzourakis, A.; Babii, N.; Scherbinska, A.; Theze, J.; Pybus, O.G.; et al. Molecular epidemiology reveals the role of war in the spread of HIV in Ukraine. Proc. Natl. Acad. Sci. USA 2018, 115, 1051–1056. [Google Scholar] [CrossRef]
- Kuzin, I.M.V.; Antonenko, J. HIV-infection in Ukraine: Information Bulletin N50; Center for Public Health: Kyiv, Ukraine, 2019. [Google Scholar]
- Magiorkinis, G.; Karamitros, T.; Vasylyeva, T.I.; Williams, L.D.; Mbisa, J.L.; Hatzakis, A.; Paraskevis, D.; Friedman, S.R. An Innovative Study Design to Assess the Community Effect of Interventions to Mitigate HIV Epidemics Using Transmission-Chain Phylodynamics. Am. J. Epidemiol. 2018, 187, 2615–2622. [Google Scholar] [CrossRef]
- Kostaki, E.G.; Frampton, D.; Paraskevis, D.; Pantavou, K.; Ferns, B.; Raffle, J.; Grant, P.; Kozlakidis, Z.; Hadjikou, A.; Pavlitina, E.; et al. Near Full-length Genomic Sequencing and Molecular Analysis of HIV-Infected Individuals in a Network-based Intervention (TRIP) in Athens, Greece: Evidence that Transmissions Occur More Frequently from those with High HIV-RNA. Curr. HIV Res. 2018, 16, 345–353. [Google Scholar] [CrossRef]
- Cunningham, S.; Ank, B.; Lewis, D.; Lu, W.; Wantman, M.; Dileanis, J.A.; Jackson, J.B.; Palumbo, P.; Krogstad, P.; Eshleman, S.H. Performance of the applied biosystems ViroSeq human immunodeficiency virus type 1 (HIV-1) genotyping system for sequence-based analysis of HIV-1 in pediatric plasma samples. J. Clin. Microbiol. 2001, 39, 1254–1257. [Google Scholar] [CrossRef]
- Pineda-Pena, A.C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gomez-Lopez, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Schmit, J.C.; Bercoff, D.P. COMET: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hue, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.; Lycett, S.; Database, U.H.D.R. Automated analysis of phylogenetic clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.S.; Pybus, O.G.; Sanders, E.J.; Albert, J.; Esbjornsson, J. Defining HIV-1 transmission clusters based on sequence data. AIDS 2017, 31, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Gelman, A.; Meng, X.L. Simulating normalizing constants: From importance sampling to bridge sampling to path sampling. Stat. Sci. 1998, 13, 163–185. [Google Scholar] [CrossRef]
- Xie, W.; Lewis, P.O.; Fan, Y.; Kuo, L.; Chen, M.H. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Syst. Biol. 2011, 60, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, A.B.; Vandamme, A.M.; Lemey, P. Quantifying differences in the tempo of human immunodeficiency virus type 1 subtype evolution. J. Virol. 2009, 83, 12917–12924. [Google Scholar] [CrossRef] [PubMed]
- Patino-Galindo, J.A.; Gonzalez-Candelas, F. The substitution rate of HIV-1 subtypes: A genomic approach. Virus Evol. 2017, 3, vex029. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, syy032. [Google Scholar] [CrossRef]
- Kuhnert, D.; Stadler, T.; Vaughan, T.G.; Drummond, A.J. Phylodynamics with Migration: A Computational Framework to Quantify Population Structure from Genomic Data. Mol. Biol. Evol. 2016, 33, 2102–2116. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Diez-Fuertes, F.; Cabello, M.; Thomson, M.M. Bayesian phylogeographic analyses clarify the origin of the HIV-1 subtype A variant circulating in former Soviet Union’s countries. Infect. Genet. Evol. 2015, 33, 197–205. [Google Scholar] [CrossRef]
- Vasylyeva, T.I.; Friedman, S.R.; Lourenco, J.; Gupta, S.; Hatzakis, A.; Pybus, O.G.; Katzourakis, A.; Smyrnov, P.; Karamitros, T.; Paraskevis, D.; et al. Reducing HIV infection in people who inject drugs is impossible without targeting recently-infected subjects. AIDS 2016, 30, 2885–2890. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]
- Grabowski, M.K.; Herbeck, J.T.; Poon, A.F.Y. Genetic Cluster Analysis for HIV Prevention. Curr. HIV/AIDS Rep. 2018, 15, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Bezemer, D.; van Sighem, A.; Lukashov, V.V.; van der Hoek, L.; Back, N.; Schuurman, R.; Boucher, C.A.; Claas, E.C.; Boerlijst, M.C.; Coutinho, R.A.; et al. Transmission networks of HIV-1 among men having sex with men in the Netherlands. AIDS 2010, 24, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.G.; Roger, M.; Routy, J.P.; Moisi, D.; Ntemgwa, M.; Matte, C.; Baril, J.G.; Thomas, R.; Rouleau, D.; Bruneau, J.; et al. High rates of forward transmission events after acute/early HIV-1 infection. J. Infect. Dis. 2007, 195, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Yerly, S.; Junier, T.; Gayet-Ageron, A.; Amari, E.B.; von Wyl, V.; Gunthard, H.F.; Hirschel, B.; Zdobnov, E.; Kaiser, L.; Swiss, H.I.V.C.S. The impact of transmission clusters on primary drug resistance in newly diagnosed HIV-1 infection. AIDS 2009, 23, 1415–1423. [Google Scholar] [CrossRef]
- Volz, E.M.; Koopman, J.S.; Ward, M.J.; Brown, A.L.; Frost, S.D. Simple epidemiological dynamics explain phylogenetic clustering of HIV from patients with recent infection. PLoS Comput. Biol. 2012, 8, e1002552. [Google Scholar] [CrossRef]
- Chalmet, K.; Staelens, D.; Blot, S.; Dinakis, S.; Pelgrom, J.; Plum, J.; Vogelaers, D.; Vandekerckhove, L.; Verhofstede, C. Epidemiological study of phylogenetic transmission clusters in a local HIV-1 epidemic reveals distinct differences between subtype B and non-B infections. BMC Infect. Dis. 2010, 10, 262. [Google Scholar] [CrossRef]
- Paraskevis, D.; Beloukas, A.; Stasinos, K.; Pantazis, N.; de Mendoza, C.; Bannert, N.; Meyer, L.; Zangerle, R.; Gill, J.; Prins, M.; et al. HIV-1 molecular transmission clusters in nine European countries and Canada: Association with demographic and clinical factors. BMC Med. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Magee, A.F.; Höhna, S.; Vasylyeva, T.I.; Leache, A.; Minin, V.N. Locally adaptive Bayesian birth-death model successfully detects slow and rapid rate shifts. BioRXiv 2019. [Google Scholar] [CrossRef]
- Vasylyeva, T. Using Molecular Epidemiology to Investigate HIV Transmission and Prevention Efforts; University of Oxford: Oxford, UK, 2018; p. 250. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total (n = 150) | Clustered (n = 22) | Non- clustered (n = 128) | Fisher’s exact test/t-test p-value *** |
|---|---|---|---|---|
| Age (mean) | 35.1 | 31.6 | 36.9 | 0.05 |
| Women (N, % *) | 46 (33.1) | 6 (27.3) | 40 (34.5) | 0.63 |
| PWID (N, %) | 52 (40.6) | 12 (54.5) | 40 (37.7) | 0.16 |
| Years since 1st injection (for PWID, mean) | 13.3 | 11.5 | 15.12 | 0.28 |
| Recently infected (N, %) | 28 (18.7) | 6 (27.3) | 22 (17.3) | 0.37 |
| HIV Viral load (mean) | 142,740.6 | 245,157.6 | 113,935.8 | 0.04 |
| HCV co-infection (N, %) | 61 (53.5) | 14 (63.6) | 47 (51.1) | 0.35 |
| HBV co-infection (N, %) | 5 (3.9) | 0 | 5 (4.9) | 0.59 |
| Syphilis co-infection (N, %) | 15 (11.6) | 3 (13.6) | 12 (11.2) | 0.72 |
| Index participants ** (N, %) | 84 (56.4) | 9 (40.9) | 75 (59.1) | 0.16 |
| Cluster | N | Year of Cluster Origin | Transm. Route | HCV | HBV | Syph-ilis | Includes Recently Infected Person | Gender | TRIP Recruit-ment Group | |
|---|---|---|---|---|---|---|---|---|---|---|
| Year | 95% HPD | |||||||||
| 1 | 2 | 2012.7 | 2010.8–2014.0 | Unk | Unk | Unk | Unk | Unk | Unk | N/A |
| 2 | 2 | 2008.0 | 2003.1–2012.9 | Drug use/sexual | Pos | Neg | Neg | Yes | Male | PTS/NPTS |
| 3 | 2 | 2014.5 | 2013.0–2015.6 | Drug use | Pos | Neg | Neg | Yes | Male | PTS |
| 4 | 2 | 2014.5 | 2012.9–2015.7 | Drug use | Neg | Neg | Neg | No | Male | PTS |
| 5 | 2 | 2014.1 | 2012.2–2015.5 | Drug use | Pos | Neg | Neg | No | Male | PTS |
| 6 | 3 | 2012.5 | 2009.7–2014.9 | Drug use | Pos | Neg | Neg/ Pos | No | Male/Female | PTS |
| 7 | 2 | 2010.7 | 2007.0–2014.0 | Drug use/sexual | Neg | Neg | Neg | Yes | Male/Female | PTS |
| 8 | 2 | 2012.9 | 2010.3–2015.1 | Sexual | Neg | Neg | Neg | Yes | Male/Female | PTS |
| 9 | 2 | 2013.9 | 2011.4–2016.0 | Drug use/sexual | Pos | Neg | Neg | No | Male | PTS |
| 10 | 2 | 2014.1 | 2012.4–2015.0 | Unk | Unk | Unk | Unk | Unk | Unk | N/A |
| 11 | 3 | 2014.2 | 2012.5–2015.5 | Sexual | Pos | Neg | Neg/ Pos | Yes | Female | PTS/NPTS |
| 12 | 2 | 2013.1 | 2010.3–2015.3 | Sexual | Neg | Neg | Neg | No | Male | PTS/NPTS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasylyeva, T.I.; Zarebski, A.; Smyrnov, P.; Williams, L.D.; Korobchuk, A.; Liulchuk, M.; Zadorozhna, V.; Nikolopoulos, G.; Paraskevis, D.; Schneider, J.; et al. Phylodynamics Helps to Evaluate the Impact of an HIV Prevention Intervention. Viruses 2020, 12, 469. https://doi.org/10.3390/v12040469
Vasylyeva TI, Zarebski A, Smyrnov P, Williams LD, Korobchuk A, Liulchuk M, Zadorozhna V, Nikolopoulos G, Paraskevis D, Schneider J, et al. Phylodynamics Helps to Evaluate the Impact of an HIV Prevention Intervention. Viruses. 2020; 12(4):469. https://doi.org/10.3390/v12040469
Chicago/Turabian StyleVasylyeva, Tetyana I., Alexander Zarebski, Pavlo Smyrnov, Leslie D. Williams, Ania Korobchuk, Mariia Liulchuk, Viktoriia Zadorozhna, Georgios Nikolopoulos, Dimitrios Paraskevis, John Schneider, and et al. 2020. "Phylodynamics Helps to Evaluate the Impact of an HIV Prevention Intervention" Viruses 12, no. 4: 469. https://doi.org/10.3390/v12040469
APA StyleVasylyeva, T. I., Zarebski, A., Smyrnov, P., Williams, L. D., Korobchuk, A., Liulchuk, M., Zadorozhna, V., Nikolopoulos, G., Paraskevis, D., Schneider, J., Skaathun, B., Hatzakis, A., Pybus, O. G., & Friedman, S. R. (2020). Phylodynamics Helps to Evaluate the Impact of an HIV Prevention Intervention. Viruses, 12(4), 469. https://doi.org/10.3390/v12040469