Unveiling the Hidden Rules of Spherical Viruses Using Point Arrays


Abstract

1. Introduction
2. Materials and Methods
2.1. Icosahedral Symmetry
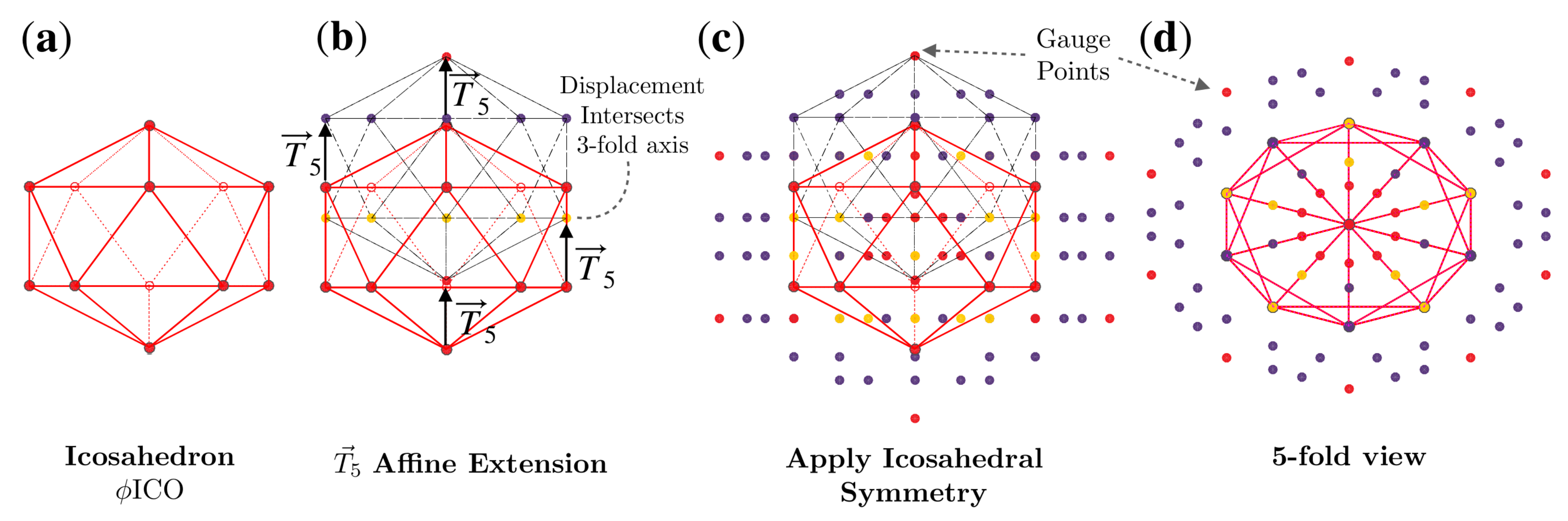
2.2. Affine Extensions
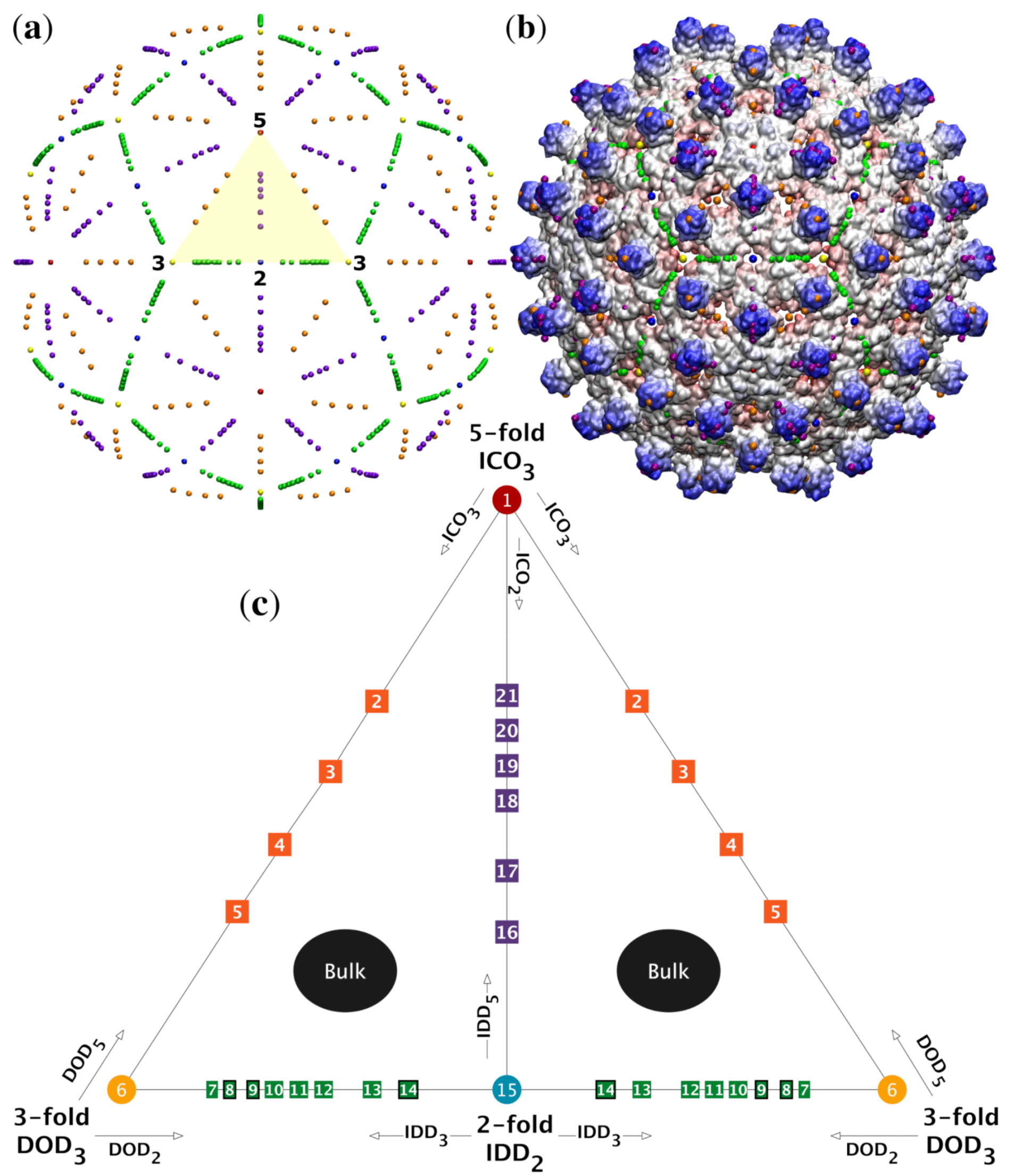
2.3. Major Features of Point Arrays
2.3.1. 55 Unique Single Point Arrays
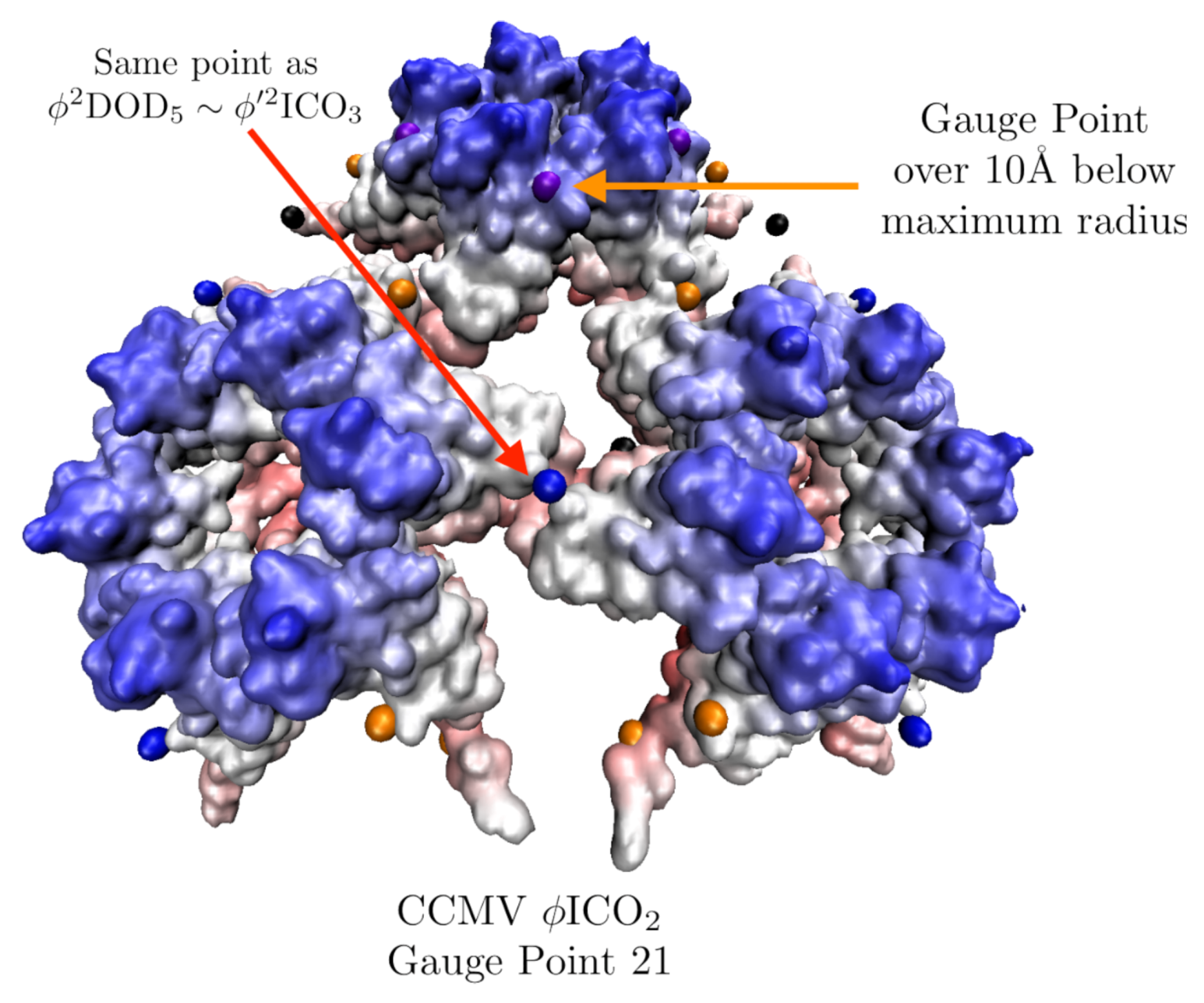
2.3.2. Gauge Points
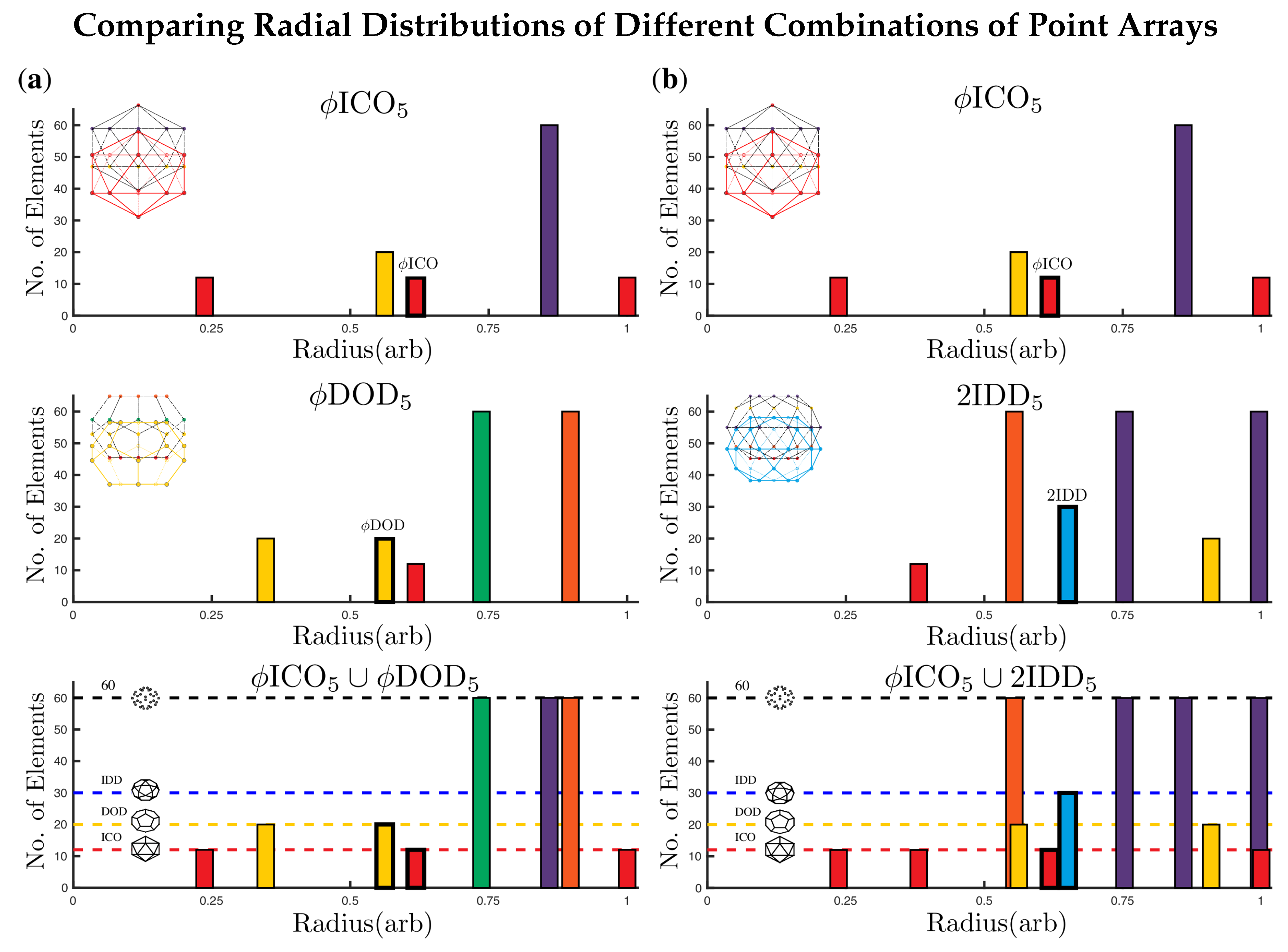
2.3.3. Sister Point Arrays
2.3.4. Double Base Point Arrays
2.3.5. Single Free Parameter
2.4. Radially Ordered Single Base Point Arrays
2.5. Point Array Fitness Algorithm
2.5.1. Identify Protruding Features of a Virus
2.5.2. Determine Gauge Point Scaling
2.5.3. Scale and Truncate Point Arrays
2.5.4. Compute RMSD from Truncated Point Arrays to the Viral Capsid Proteins
2.5.5. Determine Best Fit Point Arrays
- If a point array has a lower RMSD score by Å or more.
- Have at least one element near each protein.
- Encase the protein capsid with points above and below.
- Have a better agreement with the gauge point fits, as seen in Figure 11.
- Have more points of contact with capsid proteins, e.g., each point on the five-fold axes have at least 5 points of contact with protein surfaces. We consider this step after checking gauge point fits (d), as the number of contacts can be quite large for point arrays with bases or extensions which can considerably lower the RMSD score.
2.6. Comparison with Previous Measure
2.6.1. Gauge Point Agreement
2.6.2. Simplified RMSD Measure
2.6.3. Gauge Fixing of Truncated Point Arrays
2.6.4. Recognition of Sister Point Arrays
2.6.5. Tie Breaking Criteria
3. Results and Discussion
3.1. Virus Point Array Classification
3.2. Advantage of Sister Point Arrays
3.3. Penton Base of Adenovirus Ad3 Dodecahedron (HEV, T = 1, 4aqq)
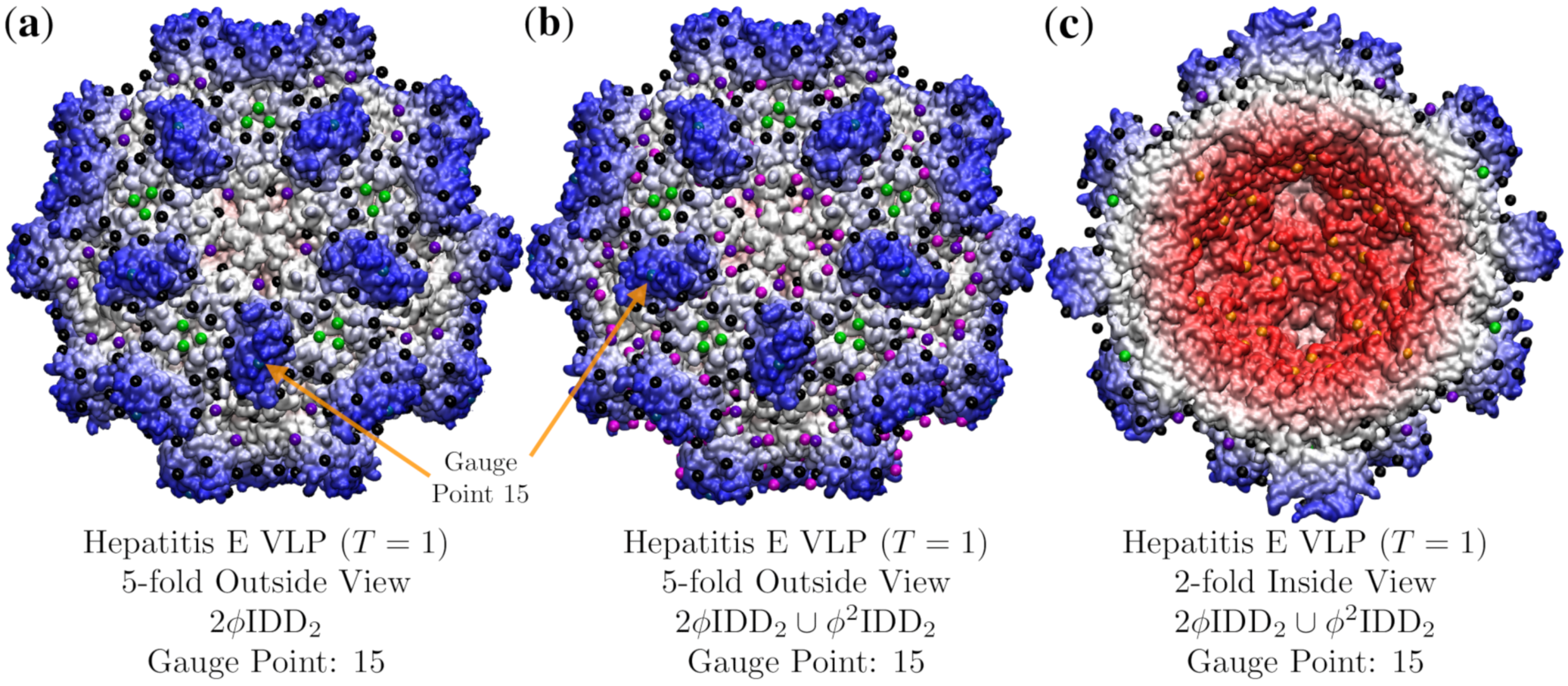
3.4. Hepatitis E VLP (HEV, T = 1, 3hag)
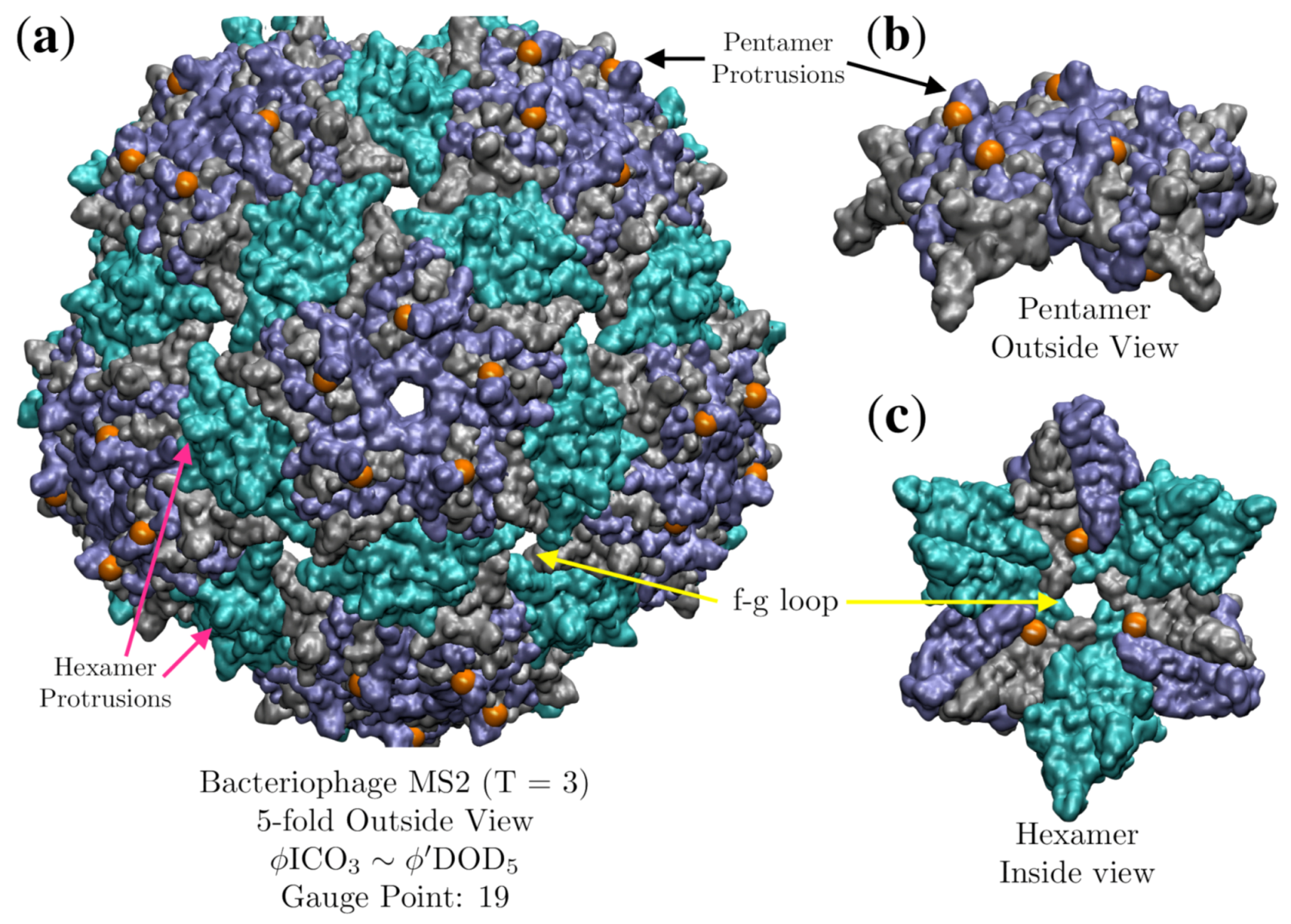
3.5. Bacteriophage MS2 (MS2, T = 3, 2ms2)
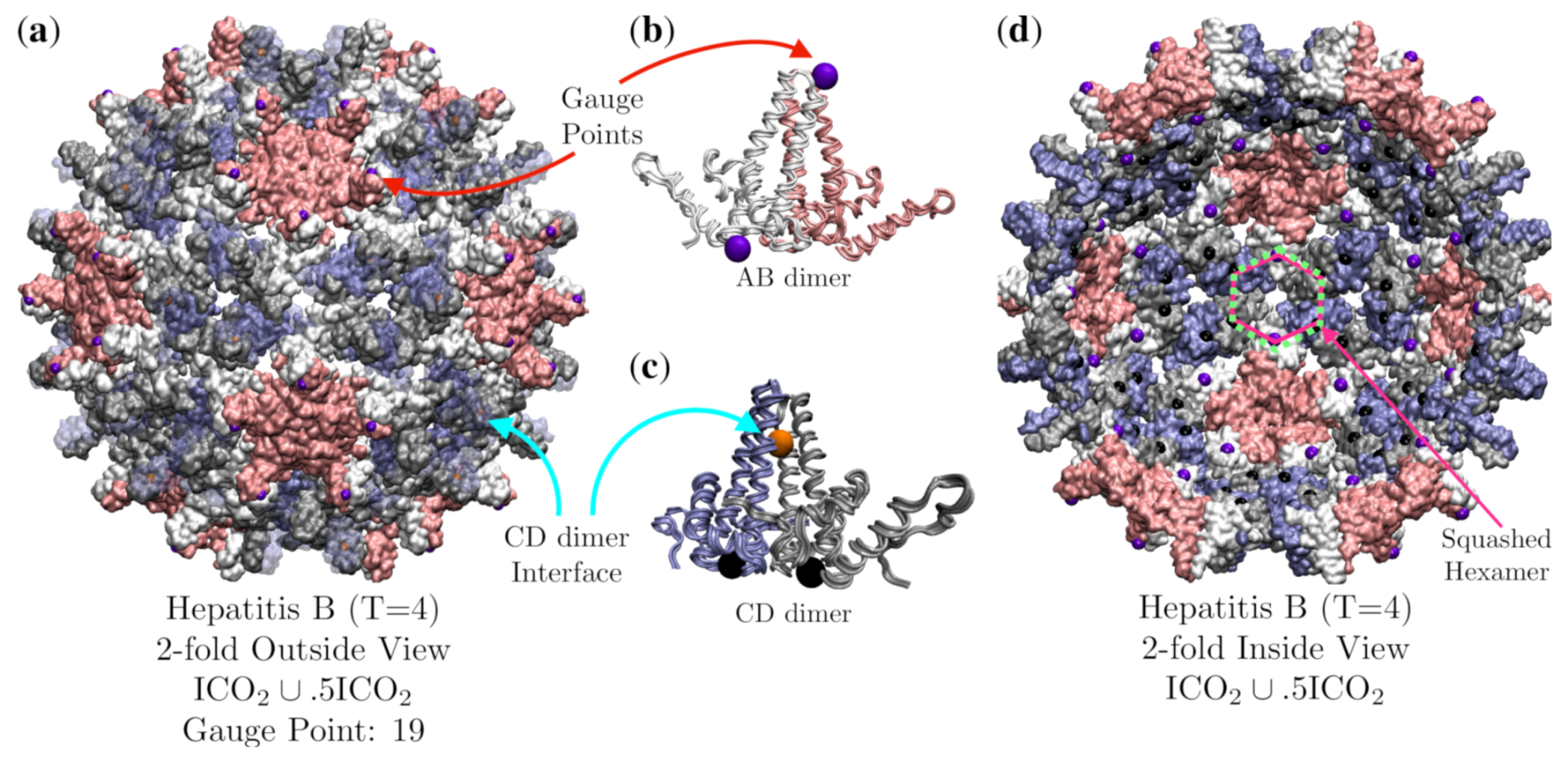
3.6. Hepatitis B (HBV, T = 4, 1qgt)
3.7. Cowpea Chlorotic Mottle Virus Maturation (CCMV, T = 3, 1cwp)
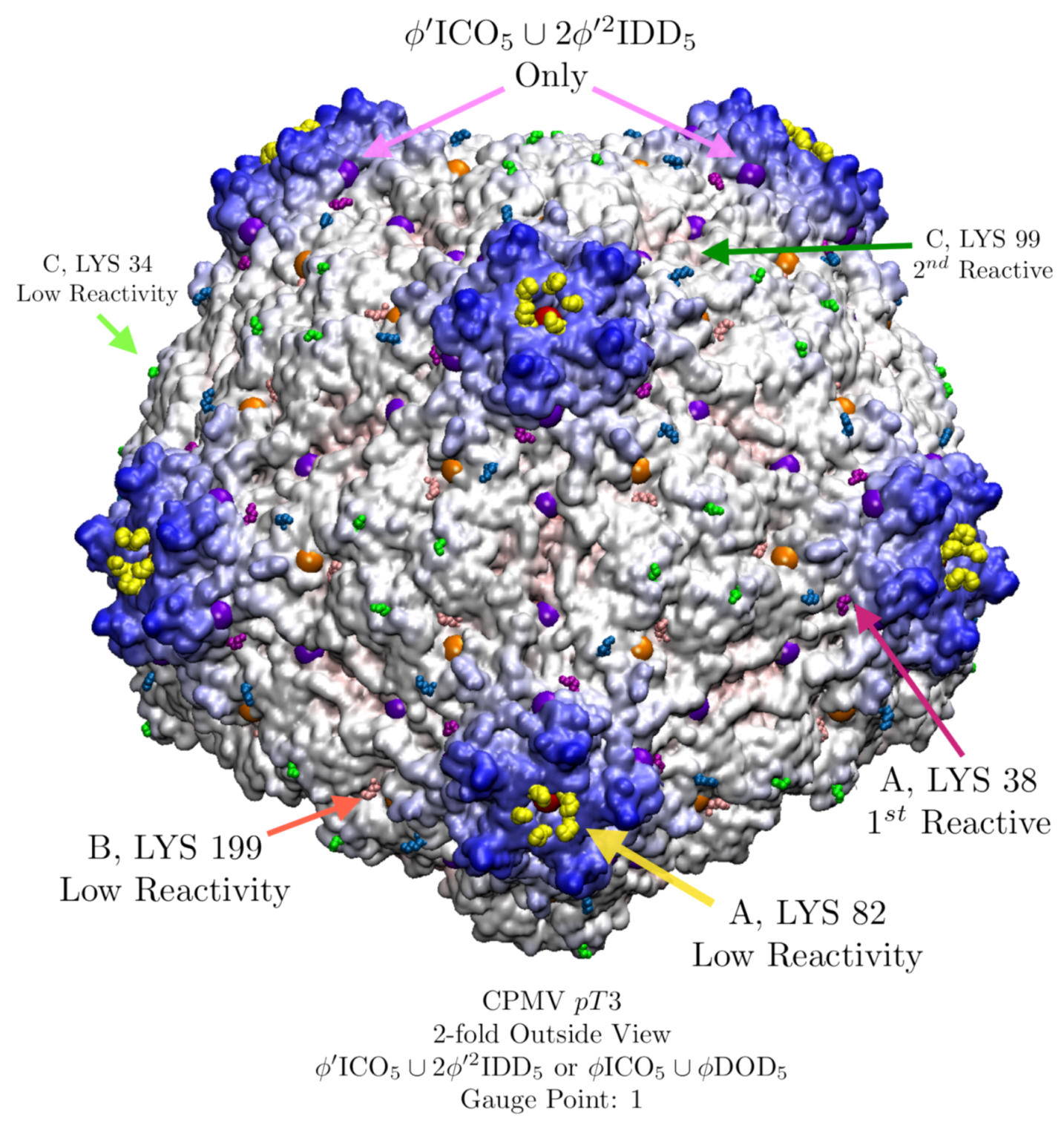
3.8. Cowpea Mosaic Virus (CPMV, pT3, 1ny7) Lysine Analysis
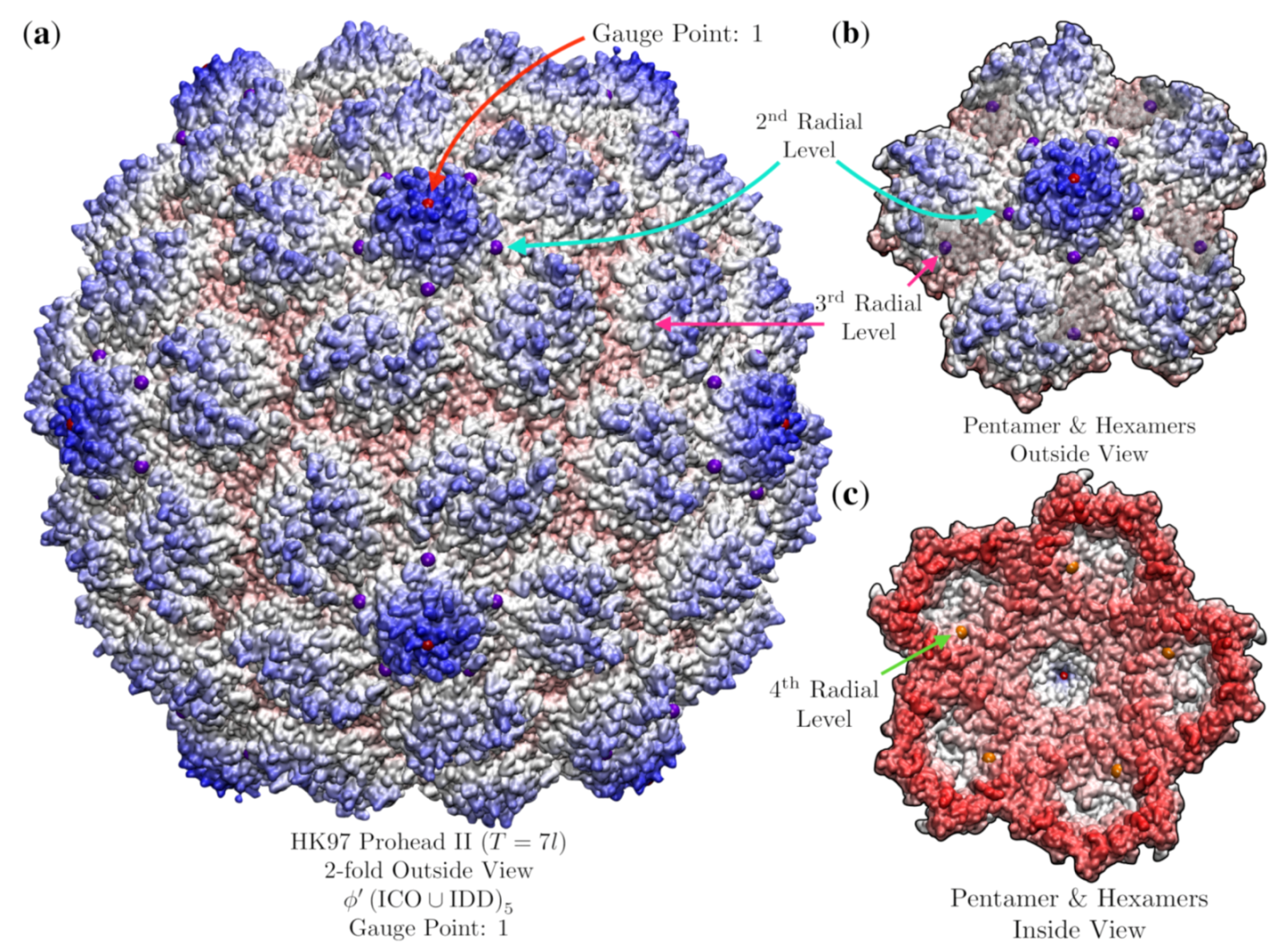
3.9. Bacteriophage HK97 Prohead II (HK97, T = , 3e8k)
3.10. Limitations of Point Arrays
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Icosahedral Rotation Matrices
Appendix A.2. Vertices of the Icosahedral Polyhedra
Appendix A.3. Worked Example of an Affine Extension
References
- Keef, T.; Twarock, R. Affine Extensions of the Icosahedral Group with Applications to the Three-dimensional Organisation of Simple Viruses. J. Math. Biol. 2009, 59, 287–313. [Google Scholar] [CrossRef]
- Keef, T.; Wardman, J.P.; Ranson, N.A.; Stockley, P.G.; Twarock, R. Structural Constraints on the Three-dimensional Geometry of Simple Viruses: Case Studies of a New Predictive Tool. Acta Crystallogr. Sect. A Found. Crystallogr. 2013, 69, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Caspar, D.L.; Klug, A. Physical Principles in the Construction of Regular Viruses. Cold Spring Harbor Symp. Quant. Biol. 1962, 27, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.P. Protruding Features of Viral Capsids are Clustered on Icosahedral Great Circles. PLoS ONE 2016, 11, 1–22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Janner, A. Form, Symmetry and Packing of Biomacro-molecules. I. Concepts and Tutorial Examples. Acta Crystallogr. Sect. A Found. Crystallogr. 2010, 66, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Janner, A. Form, Symmetry and Packing of Biomacro-molecules. II. Serotypes of Human Rhinovirus. Acta Crystallogr. Sect. A Found. Crystallogr. 2010, 66, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Janner, A. Form, Symmetry and Packing of Biomacromolecules. III. Antigenic, Receptor and Contact Binding Sites in Picornaviruses. Acta Crystallogr. Sect. A Found. Crystallogr. 2011, 67, 174–189. [Google Scholar] [CrossRef]
- Janner, A. Form, Symmetry and Packing of Biomacromolecules. IV. Filled Capsids of Cowpea, Tobacco, MS2 and Pariacoto RNA Viruses. Acta Crystallogr. Sect. A Found. Crystallogr. 2011, 67, 517–520. [Google Scholar] [CrossRef]
- Janner, A. Form, Symmetry and Packing of Biomacromolecules. V. Shells with Boundaries at anti-nodes of Resonant Vibrations in Icosahedral RNA Viruses. Acta Crystallogr. Sect. A Found. Crystallogr. 2011, 67, 521–532. [Google Scholar] [CrossRef]
- Janner, A. From an Affine Extended Icosahedral Group Towards a Toolkit for Viral Architecture. Acta Crystallogr. Sect. A Found. Crystallogr. 2013, 69, 151–163. [Google Scholar] [CrossRef]
- Zappa, E.; Dykeman, E.C.; Twarock, R. On the Subgroup Structure of the Hyperoctahedral Group in Six Dimensions. Acta Crystallogr. Sect. A Found. Adv. 2014, 70, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Zappa, E.; Dykeman, E.C.; Geraets, J.A.; Twarock, R. A Group Theoretical Approach to Structural Transitions of Icosahedral Quasicrystals and Point Arrays. J. Phys. A Math. Theor. 2016, 49, 175203. [Google Scholar] [CrossRef]
- Rochal, S.B.; Konevtsova, O.V.; Myasnikova, A.E.; Lorman, V.L. Hidden Symmetry of Small Spherical Viruses and Organization Principles in “anomalous” and Double-Shelled Capsid Nanoassemblies. Nanoscale 2016, 8, 16976–16988. [Google Scholar] [CrossRef]
- Twarock, R.; Luque, A. Structural Puzzles in Virology Solved with an Overarching Icosahedral Design Principle. Nat. Commun. 2019, 10, 4414. [Google Scholar] [CrossRef] [PubMed]
- Wardman, J.P. A Symmetry Approach to Virus Architecture. Ph.D. Thesis, University of York, York, UK, 2012. [Google Scholar]
- Wang, Q.; Kaltgrad, E.; Lin, T.; Johnson, J.E.; Finn, M.G. Natural Supramolecular Building Blocks: Wild-type Cowpea Mosaic Virus. Chem. Biol. 2002, 9, 805–811. [Google Scholar] [CrossRef]
- Chatterji, A.; Ochoa, W.F.; Paine, M.; Ratna, B.R.; Johnson, J.E.; Lin, T. New Addresses on an Addressable Virus Nanoblock: Uniquely Reactive Lys Residues on Cowpea Mosaic Virus. Chem. Biol. 2004, 11, 855–863. [Google Scholar] [CrossRef]
- Garriga, D.; Querol-Audí, J.; Abaitua, F.; Saugar, I.; Pous, J.; Verdaguer, N.; Castón, J.R.; Rodriguez, J.F. The 2.6-Angstrom Structure of Infectious Bursal Disease Virus-Derived T=1 Particles Reveals New Stabilizing Elements of the Virus Capsid. J. Virol. 2006, 80, 6895–6905. [Google Scholar] [CrossRef]
- Guu, T.S.Y.; Liu, Z.; Ye, Q.; Mata, D.A.; Li, K.; Yin, C.; Zhang, J.; Tao, Y.J. Structure of the Hepatitis E Virus-like Particle Suggests Mechanisms for Virus Assembly and Receptor Binding. Proc. Natl. Acad. Sci. USA 2009, 106, 12992–12997. [Google Scholar] [CrossRef]
- Szolajska, E.; Burmeister, W.P.; Zochowska, M.; Nerlo, B.; Andreev, I.; Schoehn, G.; Andrieu, J.P.P.; Fender, P.; Naskalska, A.; Zubieta, C.; et al. The Structural Basis for the Integrity of Adenovirus Ad3 Dodecahedron. PLoS ONE 2012, 7, 1–11. [Google Scholar] [CrossRef]
- Carrillo-Tripp, M.; Shepherd, C.M.; Borelli, I.A.; Venkataraman, S.; Lander, G.; Natarajan, P.; Johnson, J.E.; Brooks, C.L., III; Reddy, V.S. VIPERdb2: An Enhanced and Web API Enabled Relational Database for Structural Virology. Nucleic Acids Res. 2009, 37, D436–D442. [Google Scholar] [CrossRef]
- Brooks, B.; Brooks, C.L., III; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Hartman, E.C.; Jakobson, C.M.; Favor, A.H.; Lobba, M.J.; Álvarez-Benedicto, E.; Francis, M.B.; Tullman-Ercek, D. Quantitative Characterization of All Single Amino Acid Variants of a Viral Capsid-Based Drug Delivery Vehicle. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hartman, E.C.; Lobba, M.J.; Favor, A.H.; Robinson, S.A.; Francis, M.B.; Tullman-Ercek, D. Experimental Evaluation of Coevolution in a Self-Assembling Particle. Biochemistry 2019, 58, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qu, C.; Johnson, J.E.; Case, D.A. Pseudo-Atomic Models of Swollen CCMV from Cryo-Electron Microscopy Data. J. Struct. Biol. 2003, 142, 356–363. [Google Scholar] [CrossRef]
- Larson, S.B.; Day, J.; Greenwood, A.; McPherson, A. Refined Structure of Satellite Tobacco Mosaic Virus at 1.8 Å Resolution. J. Mol. Biol. 1998, 277, 37–59. [Google Scholar] [CrossRef]
- Naitow, H.; Tang, J.; Canady, M.; Wickner, R.B.; Johnson, J.E. L-A Virus at 3.4 Å Resolution Reveals Particle Architecture and mRNA Decapping Mechanism. Nat. Struct. Biol. 2002, 9, 725–728. [Google Scholar] [CrossRef]
- Tars, K.; Bundule, M.; Fridborg, K.; Liljas, L. The Crystal Structure of Bacteriophage GA and a Comparison of Bacteriophages Belonging to the Major Groups of Escherichia coli L eviviruses. J. Mol. Biol. 1997, 271, 759–773. [Google Scholar] [CrossRef]
- Golmohammadi, R.; Valegård, K.; Fridborg, K.; Liljas, L. The Refined Structure of Bacteriophage MS2 at 2.8 Å Resolution. J. Mol. Biol. 1993, 234, 620–639. [Google Scholar] [CrossRef]
- Speir, J.A.; Munshi, S.; Wang, G.; Baker, T.S.; Johnson, J.E. Structures of the Native and Swollen Forms of Cowpea Chlorotic Mottle Virus Determined by X-ray Crystallography and Cryo-Electron Microscopy. Structure 1995, 3, 63–78. [Google Scholar] [CrossRef]
- Oda, Y.; Saeki, K.; Takahashi, Y.; Maeda, T.; Naitow, H.; Tsukihara, T.; Fukuyama, K. Crystal Structure of Tobacco Necrosis Virus at 2.25 Å Resolution. J. Mol. Biol. 2000, 300, 153–169. [Google Scholar] [CrossRef]
- Lin, T.; Chen, Z.; Usha, R.; Stauffacher, C.V.; Dai, J.B.; Schmidt, T.; Johnson, J.E. The Refined Crystal Structure of Cowpea Mosaic Virus at 2.8 Å Resolution. Virology 1999, 265, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Speir, J.; Natarajan, P.; Taylor, D.; Chen, Z.; Johnson, J.E. Crystal Structure of Helicoverpa Armigera Stunt Virus. 2011; To be published. [Google Scholar]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G.W. The Crystal Structure of the Human Hepatitis B Virus Capsid. Mol. Cell 1999, 3, 771–780. [Google Scholar] [CrossRef]
- Munshi, S.; Liljas, L.; Cavarelli, J.; Bomu, W.; McKinney, B.; Reddy, V.S.; Johnson, J.E. The 2.8 Å Structure of a T = 4 Animal Virus and its Implications for Membrane Translocation of RNA. J. Mol. Biol. 1996, 261, 1–10. [Google Scholar] [CrossRef]
- Hryc, C.F.; Chen, D.H.; Afonine, P.V.; Jakana, J.; Wang, Z.; Haase-Pettingell, C.; Jiang, W.; Adams, P.D.; King, J.A.; Schmid, M.F.; et al. Accurate Model Annotation of a Near-Atomic Resolution Cryo-EM Map. Proc. Natl. Acad. Sci. USA 2017, 114, 3103–3108. [Google Scholar] [CrossRef]
- Gertsman, I.; Gan, L.; Guttman, M.; Lee, K.; Speir, J.A.; Duda, R.L.; Hendrix, R.W.; Komives, E.A.; Johnson, J.E. An unexpected twist in viral capsid maturation. Nature 2009, 458, 646–650. [Google Scholar] [CrossRef]
- Zubieta, C.; Schoehn, G.; Chroboczek, J.; Cusack, S. The Structure of the Human Adenovirus 2 Penton. Mol. Cell 2005, 17, 121–135. [Google Scholar] [CrossRef]
- Hadden, J.A.; Perilla, J.R.; Schlicksup, C.J.; Venkatakrishnan, B.; Zlotnick, A.; Schulten, K. All-atom Molecular Dynamics of the HBV Capsid Reveals Insights into Biological Function and Cryo-EM Resolution Limits. eLife 2018, 7, 1–27. [Google Scholar] [CrossRef]
- Wang, J.C.Y.; Mukhopadhyay, S.; Zlotnick, A. Geometric Defects and Icosahedral Viruses. Viruses 2018, 10, 25. [Google Scholar] [CrossRef]
- Indelicato, G.; Cermelli, P.; Salthouse, D.G.; Racca, S.; Zanzotto, G.; Twarock, R. A Crystallographic Approach to Structural Transitions in Icosahedral Viruses. J. Math. Biol. 2012, 64, 745–773. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Brooks III, C.L. Diversity and Identity of Mechanical Properties of Icosahedral Viral Capsids Studied with Elastic Network Normal Mode Analysis. J. Mol. Biol. 2005, 345, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Liljas, L.; Duda, R.L.; Tsuruta, H.; Hendrix, R.W.; Johnson, J.E. Topologically Linked Protein Rings in the Bacteriophage HK97 Capsid. Science 2000, 289, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Lošdorfer Božič, A.; Šiber, A.; Podgornik, R. Statistical Analysis of Sizes and Shapes of Virus Capsids and Their Resulting Elastic Properties. J. Biol. Phys. 2013, 39, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G. Structure of Viruses: A Short History. Q. Rev. Biophys. 2013, 46, 133–180. [Google Scholar] [CrossRef] [PubMed]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Color |
|---|---|
| 5-fold | Red |
| 5-3 GC | Orange |
| 3-fold | Yellow |
| 3-2 GC | Green |
| 2-fold | Blue |
| 2-5 GC | Purple |
| Bulk | Black |
| Origin | Teal |

| Best Fit Point Arrays with RMSD Values | ||||||
|---|---|---|---|---|---|---|
| Name | T | PA | RMSD (Å) | GP | NAU | PDBID |
| Adenovirus Ad3 Dodecahedron | 1 | 3.7 | 19 | 7 | 4aqq [20] | |
| Hepatitis E VLP | 1 | 2.8 | 15 | 26 | 3hag [19] | |
| Infectious Bursal Virus | 1 | 4.5 | 8 | 9 | 2gsy [18] | |
| STMV | 1 | 1.2 | 3 | 3 | 1a34 [26] | |
| L-A Virus | 2 | 1.4 | 4 | 3 | 1m1c [27] | |
| Bacteriophage GA | 3 | 0.2 | 2 | 2 | 1gav [28] | |
| Bacteriophage MS2 | 3 | 0.7 | 2 | 2 | 2ms2 [29] | |
| CCMV Native | 3 | 0.7 | 5 | 3 | 1cwp [30] | |
| CCMV Swollen | 3 | 2.7 | 5 | 4 | swln1 [25] | |
| Tobacco Necrosis Virus | 3 | 0.9 | 1 | 5 | 1c8n [31] | |
| Cowpea Mosaic Virus (CPMV) | 1.5 | 1 | 5 | 1ny7 [32] | ||
| Helicoverpa (HASV) | 4 | 1.1 | 19 | 6 | 3s6p [33] | |
| Hepatitis B | 4 | 1.3 | 19 | 5 | 1qgt [34] | |
| Nudaurelia Capensis Virus | 4 | 1.5 | 3 | 5 | 1ohf [35] | |
| Bacteriophage P22 Mature | 0.8 | 1 | 3 | 5uu5 [36] | ||
| HK97 Prohead II | 1.8 | 1 | 4 | 3e8k [37] | ||
| CCMV Swollen | ||||
|---|---|---|---|---|
| PA | RMSD | GP | NAU | Notes |
| 1.9 | 5 | 3 | ||
| 2.2 | 21 | 6 | excluded | |
| 2.3 | 21 | 7 | excluded | |
| 2.6 | 21 | 8 | excluded | |
| 2.7 | 5 | 4 | ||
| CPMV (pT = 3) LYS Reactivity Comparison | ||||
|---|---|---|---|---|
| Residue | Reactivity | or | Accessibility | Naive |
| Prediction | ||||
| A LYS 82 | Low | X/X | Solvent: +, Sterics: − | Good |
| A LYS 38 | Highest | X/+ | Solvent: −, Sterics: + | Poor |
| B LYS 199 | Low | X/X | Solvent: +, Sterics: − | Good |
| C LYS 34 | Low | +/+ | Solvent: +, Sterics: − | Good |
| C LYS 99 | Second | +/+ | Solvent: −, Sterics: + | Poor |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, D.P. Unveiling the Hidden Rules of Spherical Viruses Using Point Arrays. Viruses 2020, 12, 467. https://doi.org/10.3390/v12040467
Wilson DP. Unveiling the Hidden Rules of Spherical Viruses Using Point Arrays. Viruses. 2020; 12(4):467. https://doi.org/10.3390/v12040467
Chicago/Turabian StyleWilson, David P. 2020. "Unveiling the Hidden Rules of Spherical Viruses Using Point Arrays" Viruses 12, no. 4: 467. https://doi.org/10.3390/v12040467
APA StyleWilson, D. P. (2020). Unveiling the Hidden Rules of Spherical Viruses Using Point Arrays. Viruses, 12(4), 467. https://doi.org/10.3390/v12040467