HIV Reactivation in Latently Infected Cells with Virological Synapse-Like Cell Contact

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Cell Coculture in the Presence or Absence of Cell-to-Cell Contacts
2.3. Monitoring of HIV-1 p24 Production and TNF-α Secretion
2.4. Flow Cytometry (FCM)
2.5. Fluorescence Microscopy
2.6. Preparation and Analysis of Cell Surface Fractions
2.7. Isobaric Tag for Relative and Absolute Quantitation (iTRAQ) Analysis of Cell Surface Fractions
3. Results
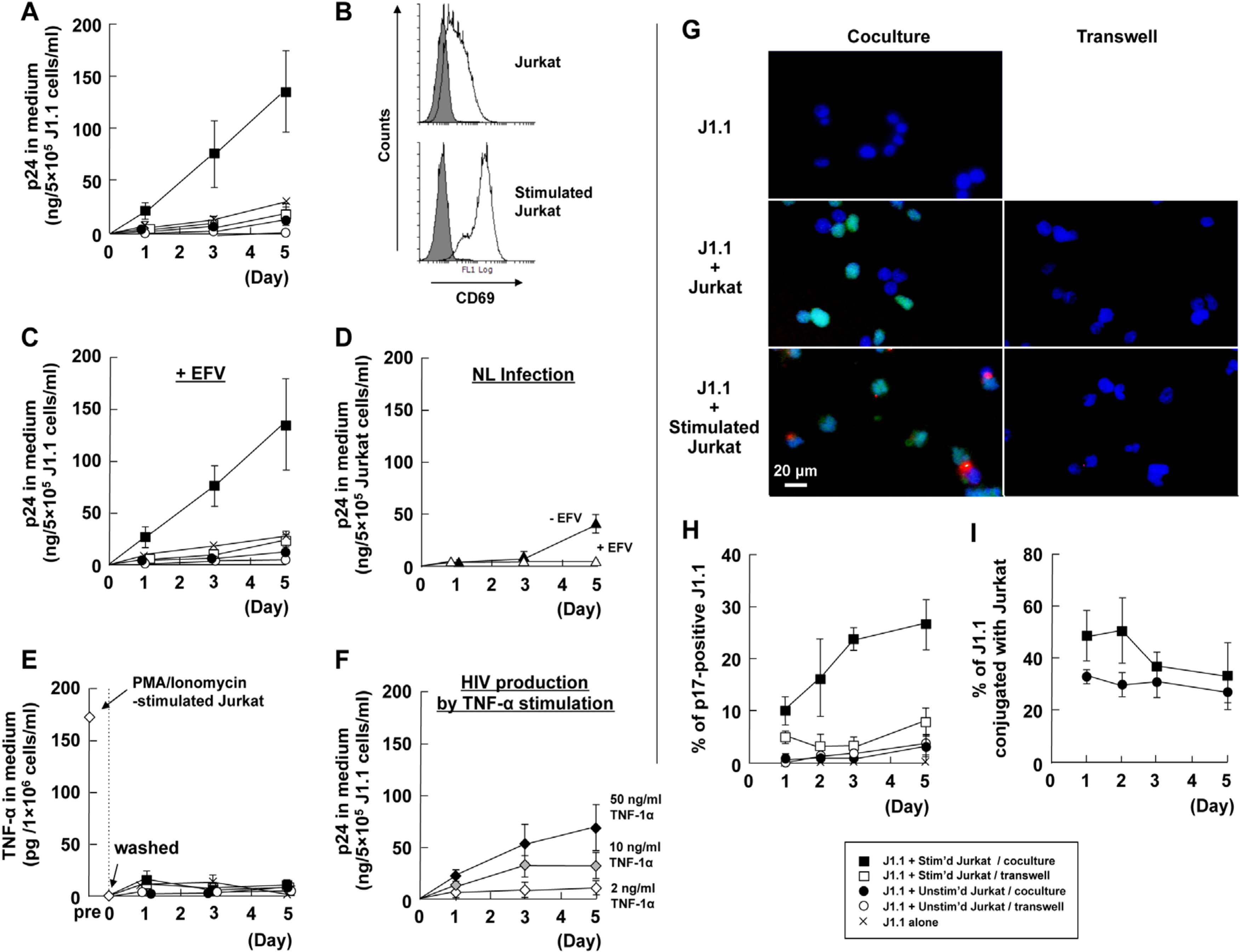
3.1. Cellular Contact with Stimulated T Cells Efficiently Reactivates Latent HIV-1 Provirus
3.2. LFA-1-ICAM-1 Interaction is Involved in the Cell-to-Cell Contact, Leading to Latent HIV-1 Reactivation
3.3. Cell-to-Cell Contact of Latently Infected T Cells and Stimulated T Cells forms Virological Synapse-Like Structure
3.4. Identification of Molecules on Stimulated T Cells which are Involved in Reactivation
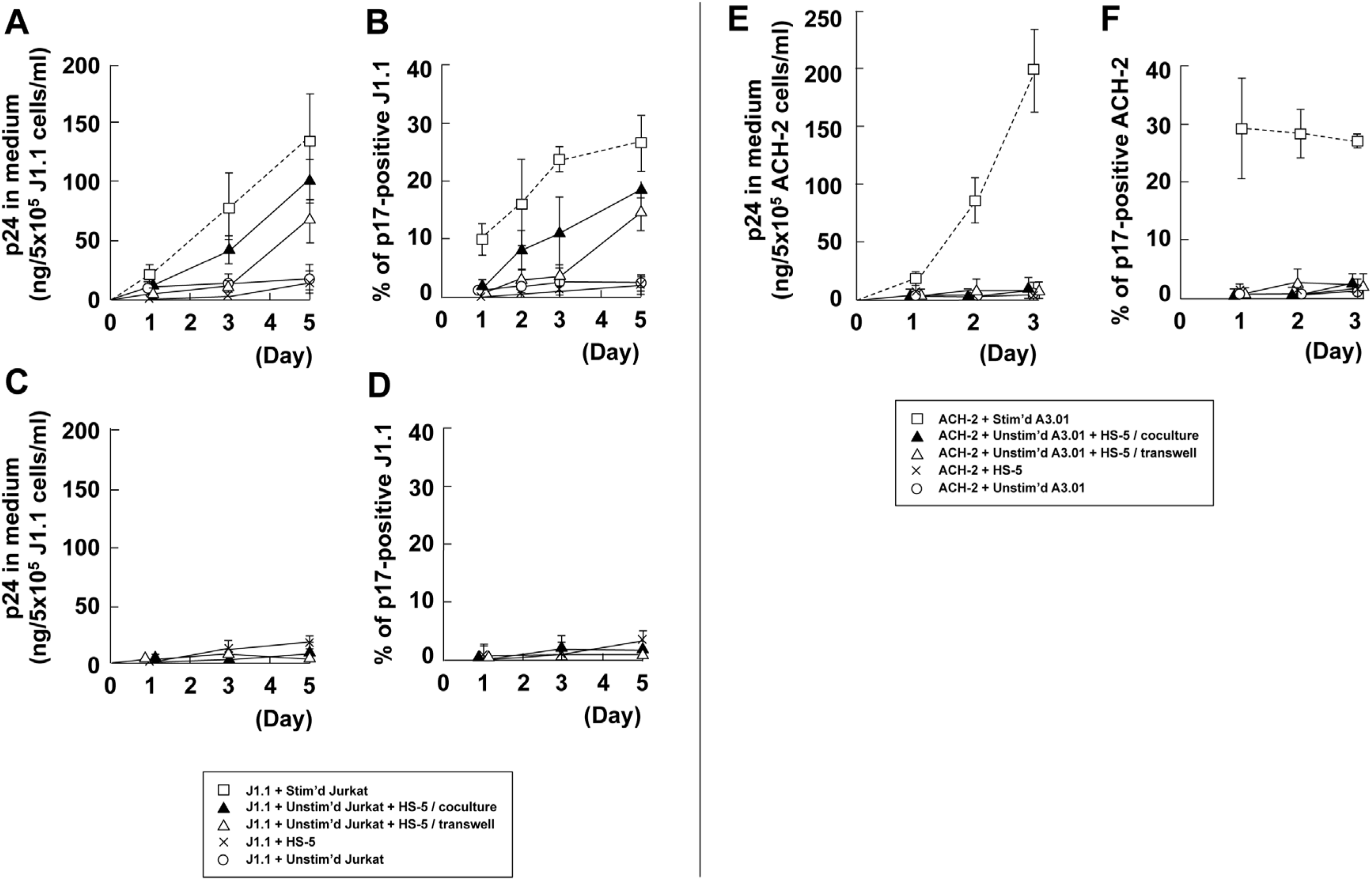
3.5. Stromal Cells Contribute to Efficient Reactivation of Latent HIV-1 in T Cell Cocultures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Fauci, A.S. HIV reservoirs: Pathogenesis and obstacles to viral eradication and cure. AIDS 2012, 26, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In Vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of HIV in Gut-Associated Lymphoid Tissue despite Long-Term Antiretroviral Therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef]
- Alexaki, A.; Wigdahl, B. HIV-1 Infection of Bone Marrow Hematopoietic Progenitor Cells and Their Role in Trafficking and Viral Dissemination. PLoS Pathog. 2008, 4, e1000215. [Google Scholar] [CrossRef]
- McNamara, L.A.; Ganesh, J.A.; Collins, K.L. Latent HIV-1 Infection Occurs in Multiple Subsets of Hematopoietic Progenitor Cells and Is Reversed by NF-B Activation. J. Virol. 2012, 86, 9337–9350. [Google Scholar] [CrossRef]
- Schneider, M.; Tigges, B.; Meggendorfer, M.; Helfer, M.; Ziegenhain, C.; Brack-Werner, R. A new model for post-integration latency in macroglial cells to study HIV-1 reservoirs of the brain. AIDS 2015, 29, 1147–1159. [Google Scholar] [CrossRef]
- De Jong, M.D.; de Boer, R.J.; de Wolf, F.; Foudraine, N.A.; Boucher, C.A.B.; Goudsmit, J.; Lange, J.M.A. Overshoot of HIV-1 viraemia after early discontinuation of antiretroviral treatment. AIDS 1997, 11, F79–F84. [Google Scholar] [CrossRef]
- Chun, T.-W.; Fauci, A.S. Latent reservoirs of HIV: Obstacles to the eradication of virus. Proc. Natl. Acad. Sci. USA 1999, 96, 10958–10961. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for Viral Persistence in the Presence of Antiviral Immune Responses and Antiretroviral Therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Engel, D.; Mizell, S.B.; Hallahan, C.W.; Fischette, M.; Park, S.; Davey, R.T.; Dybul, M.; Kovacs, J.A.; Metcalf, J.A.; et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat. Med. 1999, 5, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Emery, S. Latent reservoirs of HIV infection: Flushing with IL-2? Nat. Med. 1999, 5, 611–612. [Google Scholar] [CrossRef]
- Zhong, P.; Agosto, L.M.; Ilinskaya, A.; Dorjbal, B.; Truong, R.; Derse, D.; Uchil, P.D.; Heidecker, G.; Mothes, W. Cell-to-Cell Transmission Can Overcome Multiple Donor and Target Cell Barriers Imposed on Cell-Free HIV. PLoS ONE 2013, 8, e53138. [Google Scholar] [CrossRef]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 Cell to Cell Transfer across an Env-induced, Actin-dependent Synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef]
- Chen, P.; Hubner, W.; Spinelli, M.A.; Chen, B.K. Predominant Mode of Human Immunodeficiency Virus Transfer between T Cells Is Mediated by Sustained Env-Dependent Neutralization-Resistant Virological Synapses. J. Virol. 2007, 81, 12582–12595. [Google Scholar] [CrossRef]
- Hübner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.S.; Li, X.-D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3D Video Microscopy of HIV Transfer Across T Cell Virological Synapses. Science 2009, 323, 1743–1747. [Google Scholar] [CrossRef]
- Rudnicka, D.; Feldmann, J.; Porrot, F.; Wietgrefe, S.; Guadagnini, S.; Prevost, M.-C.; Estaquier, J.; Haase, A.T.; Sol-Foulon, N.; Schwartz, O. Simultaneous Cell-to-Cell Transmission of Human Immunodeficiency Virus to Multiple Targets through Polysynapses. J. Virol. 2009, 83, 6234–6246. [Google Scholar] [CrossRef]
- Llewellyn, G.N.; Hogue, I.B.; Grover, J.R.; Ono, A. Nucleocapsid Promotes Localization of HIV-1 Gag to Uropods That Participate in Virological Synapses between T Cells. PLoS Pathog. 2010, 6, e1001167. [Google Scholar] [CrossRef]
- Wang, L.; Eng, E.T.; Law, K.; Gordon, R.E.; Rice, W.J.; Chen, B.K. Visualization of HIV T Cell Virological Synapses and Virus-Containing Compartments by Three-Dimensional Correlative Light and Electron Microscopy. J. Virol. 2017, 91, e01605–e01616. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.H.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R. DC-SIGN, a Dendritic Cell-Specific HIV-1-Binding Protein that Enhances trans-Infection of T Cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- McDonald, D.; Wu, L.; Bohks, S.M.; KewalRamani, V.N.; Unutmaz, D.; Hope, T.J. Recruitment of HIV and Its Receptors to Dendritic Cell-T Cell Junctions. Science 2003, 300, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D. Dendritic Cells and HIV-1 Trans-Infection. Viruses 2010, 2, 1704–1717. [Google Scholar] [CrossRef]
- Vasiliver-Shamis, G.; Tuen, M.; Wu, T.W.; Starr, T.; Cameron, T.O.; Thomson, R.; Kaur, G.; Liu, J.; Visciano, M.L.; Li, H.; et al. Human Immunodeficiency Virus Type 1 Envelope gp120 Induces a Stop Signal and Virological Synapse Formation in Noninfected CD4+ T Cells. J. Virol. 2008, 82, 9445–9457. [Google Scholar] [CrossRef]
- Vasiliver-Shamis, G.; Cho, M.W.; Hioe, C.E.; Dustin, M.L. Human Immunodeficiency Virus Type 1 Envelope gp120-Induced Partial T-Cell Receptor Signaling Creates an F-Actin-Depleted Zone in the Virological Synapse. J. Virol. 2009, 83, 11341–11355. [Google Scholar] [CrossRef] [PubMed]
- Vasiliver-Shamis, G.; Dustin, M.; Hioe, C. HIV-1 Virological Synapse is not Simply a Copycat of the Immunological Synapse. Viruses 2010, 2, 1239–1260. [Google Scholar] [CrossRef]
- Folks, T.; Justement, J.; Kinter, A.; Dinarello, C.; Fauci, A. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987, 238, 800–802. [Google Scholar] [CrossRef]
- Poli, G.; Kinter, A.L.; Fauci, A.S. Interleukin 1 induces expression of the human immunodeficiency virus alone and in synergy with interleukin 6 in chronically infected U1 cells: Inhibition of inductive effects by the interleukin 1 receptor antagonist. Proc. Natl. Acad. Sci. USA 1994, 91, 108–112. [Google Scholar] [CrossRef]
- Scripture-Adams, D.D.; Brooks, D.G.; Korin, Y.D.; Zack, J.A. Interleukin-7 Induces Expression of Latent Human Immunodeficiency Virus Type 1 with Minimal Effects on T-Cell Phenotype. J. Virol. 2002, 76, 13077–13082. [Google Scholar] [CrossRef]
- Managlia, E.Z.; Landay, A.; Al-Harthi, L. Interleukin-7 induces HIV replication in primary naive T cells through a nuclear factor of activated T cell (NFAT)-dependent pathway. Virology 2006, 350, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.A.; Greene, W.C. Regulation of HIV-1 latency by T-cell activation. Cytokine 2007, 39, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Devadas, K.; Hardegen, N.J.; Wahl, L.M.; Hewlett, I.K.; Clouse, K.A.; Yamada, K.M.; Dhawan, S. Mechanisms for Macrophage-Mediated HIV-1 Induction. J. Immunol. 2004, 173, 6735–6744. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Koya, Y.; Saitoh, T.; Saitoh, Y.; Shimizu, S.; Ohba, K.; Yamamoto, N.; Yamaoka, S.; Yamamoto, N. Efficient induction of HIV-1 replication in latently infected cells through contact with CD4+ T cells: Involvement of NF-κB activation. Virology 2007, 361, 325–334. [Google Scholar] [CrossRef]
- Van der Sluis, R.M.; van Montfort, T.; Pollakis, G.; Sanders, R.W.; Speijer, D.; Berkhout, B.; Jeeninga, R.E. Dendritic Cell-induced Activation of Latent HIV-1 Provirus in Actively Proliferating Primary T Lymphocytes. PLoS Pathog. 2013, 9, e1003259. [Google Scholar] [CrossRef]
- Borghi, M.O.; Panzeri, P.; Shattock, R.; Sozzani, S.; Dobrina, A.; Meroni, P.L. Interaction between chronically HIV-infected promonocytic cells and human umbilical vein endothelial cells: Role of proinflammatory cytokines and chemokines in viral expression modulation. Clin. Exp. Immunol. 2000, 120, 93–100. [Google Scholar] [CrossRef]
- Gilles, P.N.; Lathey, J.L.; Spector, S.A. Replication of macrophage-tropic and T-cell-tropic strains of human immunodeficiency virus type 1 is augmented by macrophage-endothelial cell contact. J. Virol. 1995, 69, 2133–2139. [Google Scholar] [CrossRef]
- Fan, S.; Hsia, K.; Edgington, T. Upregulation of human immunodeficiency virus-1 in chronically infected monocytic cell line by both contact with endothelial cells and cytokines. Blood 1994, 84, 1567–1572. [Google Scholar] [CrossRef]
- Shen, A.; Baker, J.J.; Scott, G.L.; Davis, Y.P.; Ho, Y.-Y.; Siliciano, R.F. Endothelial Cell Stimulation Overcomes Restriction and Promotes Productive and Latent HIV-1 Infection of Resting CD4+ T Cells. J. Virol. 2013, 87, 9768–9779. [Google Scholar] [CrossRef]
- Mueller, S.N.; Germain, R.N. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat. Rev. Immunol. 2009, 9, 618–629. [Google Scholar] [CrossRef]
- Turley, S.J.; Fletcher, A.L.; Elpek, K.G. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat. Rev. Immunol. 2010, 10, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Gerlach, S.L.; Wu, C.; Khurana, N.; Swientoniewski, L.T.; Abdel-Mageed, A.B.; Li, J.; Braun, S.E.; Mondal, D. Mesenchymal stem cells are attracted to latent HIV-1-infected cells and enable virus reactivation via a non-canonical PI3K-NFκB signaling pathway. Sci. Rep. 2018, 8, 14702. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.L.; Rowe, T.; Justement, J.S.; Butera, S.T.; June, C.H.; Folks, T.M. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J. Immunol. 1991, 147, 3145–3148. [Google Scholar]
- Clouse, K.A.; Powell, D.; Washington, I.; Poli, G.; Strebel, K.; Farrar, W.; Barstad, P.; Kovacs, J.; Fauci, A.S.; Folks, T.M. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 1989, 142, 431–438. [Google Scholar] [PubMed]
- Kaplan, A.H.; Manchester, M.; Swanstrom, R. The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J. Virol. 1994, 68, 6782–6786. [Google Scholar] [CrossRef]
- Katagiri, K.; Hattori, M.; Minato, N.; Irie, S.-K.; Takatsu, K.; Kinashi, T. Rap1 Is a Potent Activation Signal for Leukocyte Function-Associated Antigen 1 Distinct from Protein Kinase C and Phosphatidylinositol-3-OH Kinase. Mol. Cell. Biol. 2000, 20, 1956–1969. [Google Scholar] [CrossRef] [PubMed]
- Terho, P. Flowing Software. Available online: http://www.flowingsoftware.com (accessed on 8 April 2020).
- Berro, R.; de la Fuente, C.; Klase, Z.; Kehn, K.; Parvin, L.; Pumfery, A.; Agbottah, E.; Vertes, A.; Nekhai, S.; Kashanchi, F. Identifying the Membrane Proteome of HIV-1 Latently Infected Cells. J. Biol. Chem. 2007, 282, 8207–8218. [Google Scholar] [CrossRef]
- Suyama, M.; Daikoku, E.; Goto, T.; Sano, K.; Morikawa, Y. Reactivation from latency displays HIV particle budding at plasma membrane, accompanying CD44 upregulation and recruitment. Retrovirology 2009, 6, 63. [Google Scholar] [CrossRef]
- Weber, K.S.; York, M.R.; Springer, T.A.; Klickstein, L.B. Characterization of lymphocyte function-associated antigen 1 (LFA-1)-deficient T cell lines: The alphaL and beta2 subunits are interdependent for cell surface expression. J. Immunol. 1997, 158, 273–279. [Google Scholar]
- Groot, F.; Kuijpers, T.W.; Berkhout, B.; de Jong, E.C. Dendritic cell-mediated HIV-1 transmission to T cells of LAD-1 patients is impaired due to the defect in LFA-1. Retrovirology 2006, 3, 1–9. [Google Scholar] [CrossRef]
- Jolly, C.; Mitar, I.; Sattentau, Q.J. Adhesion Molecule Interactions Facilitate Human Immunodeficiency Virus Type 1-Induced Virological Synapse Formation between T Cells. J. Virol. 2007, 81, 13916–13921. [Google Scholar] [CrossRef] [PubMed]
- Starling, S.; Jolly, C. LFA-1 Engagement Triggers T Cell Polarization at the HIV-1 Virological Synapse. J. Virol. 2016, 90, 9841–9854. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Freed, E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 2001, 98, 13925–13930. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.; Weclewicz, K.; Hewson, R.; Suomalainen, M. Human Immunodeficiency Virus Type 1 Assembly and Lipid Rafts: Pr55gag Associates with Membrane Domains That Are Largely Resistant to Brij98 but Sensitive to Triton X-100. J. Virol. 2003, 77, 4805–4817. [Google Scholar] [CrossRef]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag Induces the Coalescence of Clustered Lipid Rafts and Tetraspanin-Enriched Microdomains at HIV-1 Assembly Sites on the Plasma Membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar] [CrossRef]
- Nydegger, S.; Khurana, S.; Krementsov, D.N.; Foti, M.; Thali, M. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J. Cell Biol. 2006, 173, 795–807. [Google Scholar] [CrossRef]
- Krementsov, D.N.; Rassam, P.; Margeat, E.; Roy, N.H.; Schneider-Schaulies, J.; Milhiet, P.-E.; Thali, M. HIV-1 Assembly Differentially Alters Dynamics and Partitioning of Tetraspanins and Raft Components. Traffic 2010, 11, 1401–1414. [Google Scholar] [CrossRef]
- Len, A.C.L.; Starling, S.; Shivkumar, M.; Jolly, C. HIV-1 Activates T Cell Signaling Independently of Antigen to Drive Viral Spread. Cell Rep. 2017, 18, 1062–1074. [Google Scholar] [CrossRef]
- Schneider, U.; Schwenk, H.-U.; Bornkamm, G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J. Cancer 1977, 19, 621–626. [Google Scholar] [CrossRef]
- Weiss, A.; Stobo, J.D. Requirement for the coexpression of T3 and the T cell antigen receptor on a malignant human T cell line. J. Exp. Med. 1984, 160, 1284–1299. [Google Scholar] [CrossRef]
- Jóźwik, A.; Soroczyńska, M.; Witkowski, J.M.; Bryl, E. CD3 receptor modulation in Jurkat leukemic cell line. Folia Histochem. Cytobiol. 2004, 42, 41–43. [Google Scholar] [CrossRef]
- Miller, L.J.; Springer, T.A. Biosynthesis and glycosylation of p150,95 and related leukocyte adhesion proteins. J. Immunol. 1987, 139, 842–847. [Google Scholar] [PubMed]
- Roecklein, B.; Torok-Storb, B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood 1995, 85, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Graf, L.; Iwata, M.; Torok-Storb, B. Gene expression profiling of the functionally distinct human bone marrow stromal cell lines HS-5 and HS-27a. Blood 2002, 100, 1509–1511. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Sattentau, Q.J. Human Immunodeficiency Virus Type 1 Virological Synapse Formation in T Cells Requires Lipid Raft Integrity. J. Virol. 2005, 79, 12088–12094. [Google Scholar] [CrossRef]
- Jolly, C.; Sattentau, Q.J. Human Immunodeficiency Virus Type 1 Assembly, Budding, and Cell-Cell Spread in T Cells Take Place in Tetraspanin-Enriched Plasma Membrane Domains. J. Virol. 2007, 81, 7873–7884. [Google Scholar] [CrossRef]
- Feldmann, J.; Schwartz, O. HIV-1 Virological Synapse: Live Imaging of Transmission. Viruses 2010, 2, 1666–1680. [Google Scholar] [CrossRef]
- Chan, S.M.; Ermann, J.; Su, L.; Fathman, C.G.; Utz, P.J. Protein microarrays for multiplex analysis of signal transduction pathways. Nat. Med. 2004, 10, 1390–1396. [Google Scholar] [CrossRef]
- Tu, X.; Huang, A.; Bae, D.; Slaughter, N.; Whitelegge, J.; Crother, T.; Bickel, P.E.; Nel, A. Proteome Analysis of Lipid Rafts in Jurkat Cells Characterizes a Raft Subset That Is Involved in NF-κB Activation. J. Proteome Res. 2004, 3, 445–454. [Google Scholar] [CrossRef]
- Peirce, M.J.; Wait, R.; Begum, S.; Saklatvala, J.; Cope, A.P. Expression Profiling of Lymphocyte Plasma Membrane Proteins. Mol. Cell. Proteom. 2004, 3, 56–65. [Google Scholar] [CrossRef]
- Saleh, S.; Solomon, A.; Wightman, F.; Xhilaga, M.; Cameron, P.U.; Lewin, S.R. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: A novel model of HIV-1 latency. Blood 2007, 110, 4161–4164. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.; Wightman, F.; Ramanayake, S.; Alexander, M.; Kumar, N.; Khoury, G.; Pereira, C.; Purcell, D.; Cameron, P.U.; Lewin, S.R. Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology 2011, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Sherrill-Mix, S.; Lewinski, M.K.; Famiglietti, M.; Bosque, A.; Malani, N.; Ocwieja, K.E.; Berry, C.C.; Looney, D.; Shan, L.; Agosto, L.M.; et al. HIV latency and integration site placement in five cell-based models. Retrovirology 2013, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, S.D.; Cameron, P.U. Human Immunodeficiency Virus (HIV)-1 Integration Sites in Viral Latency. Curr. HIV AIDS Rep. 2015, 12, 88–96. [Google Scholar] [CrossRef]
- Park, J.; Lim, C.H.; Ham, S.; Kim, S.S.; Choi, B.-S.; Roh, T.-Y. Genome-wide analysis of histone modifications in latently HIV-1 infected T cells. AIDS 2014, 28, 1719–1728. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okutomi, T.; Minakawa, S.; Hirota, R.; Katagiri, K.; Morikawa, Y. HIV Reactivation in Latently Infected Cells with Virological Synapse-Like Cell Contact. Viruses 2020, 12, 417. https://doi.org/10.3390/v12040417
Okutomi T, Minakawa S, Hirota R, Katagiri K, Morikawa Y. HIV Reactivation in Latently Infected Cells with Virological Synapse-Like Cell Contact. Viruses. 2020; 12(4):417. https://doi.org/10.3390/v12040417
Chicago/Turabian StyleOkutomi, Toshiki, Satoko Minakawa, Riku Hirota, Koko Katagiri, and Yuko Morikawa. 2020. "HIV Reactivation in Latently Infected Cells with Virological Synapse-Like Cell Contact" Viruses 12, no. 4: 417. https://doi.org/10.3390/v12040417
APA StyleOkutomi, T., Minakawa, S., Hirota, R., Katagiri, K., & Morikawa, Y. (2020). HIV Reactivation in Latently Infected Cells with Virological Synapse-Like Cell Contact. Viruses, 12(4), 417. https://doi.org/10.3390/v12040417