Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death



Abstract
1. Introduction
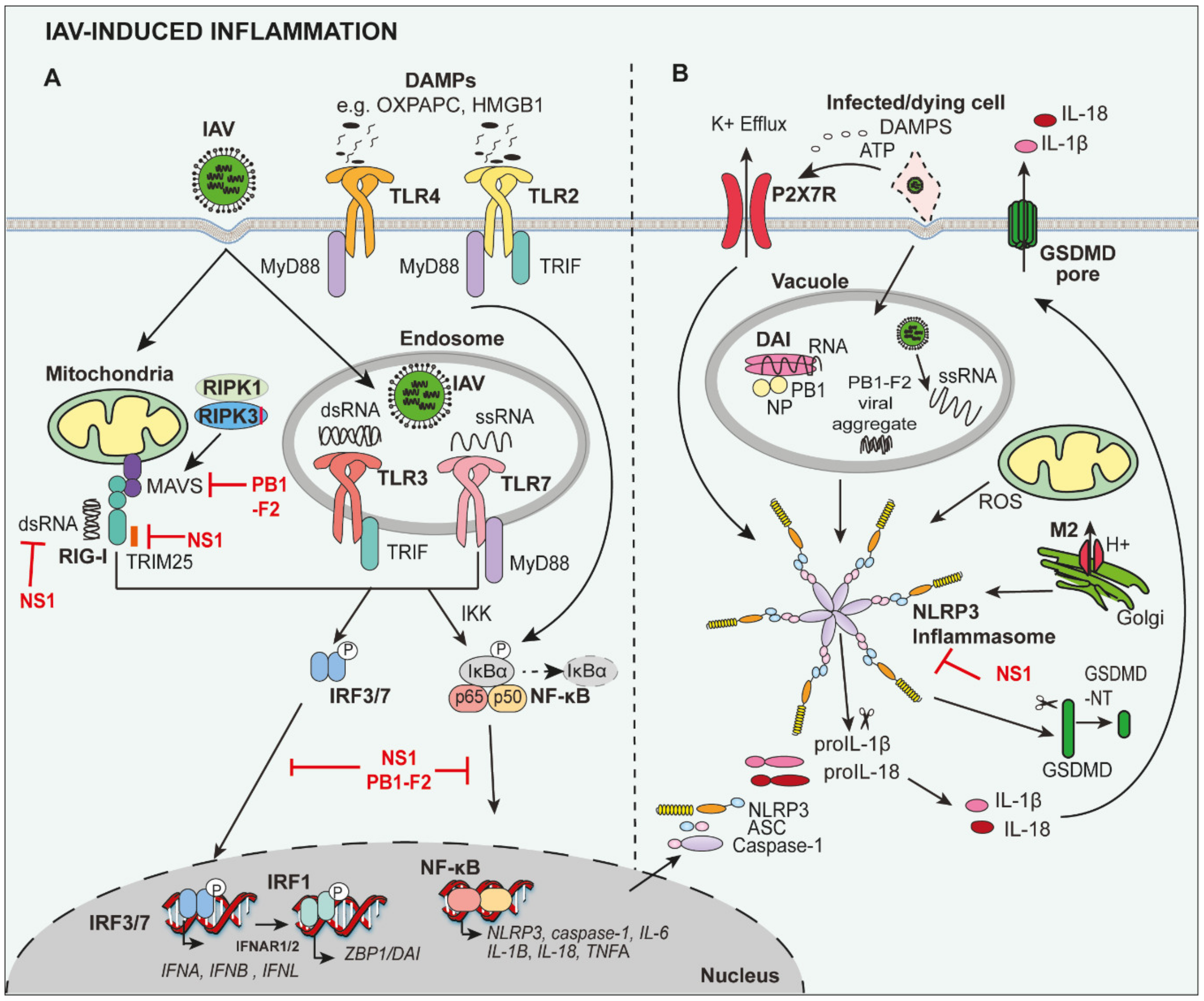
2. Induction of Inflammation during IAV Infection
3. Inflammatory Responses in Mediating Host Defence and Promoting Disease during Severe IAV Infection
3.1. PRRs Mediate Protective and Detrimental Inflammation
3.2. Pro-Inflammatory Cytokines and the ‘Cytokine Storm’
3.3. Cellular Immune Response
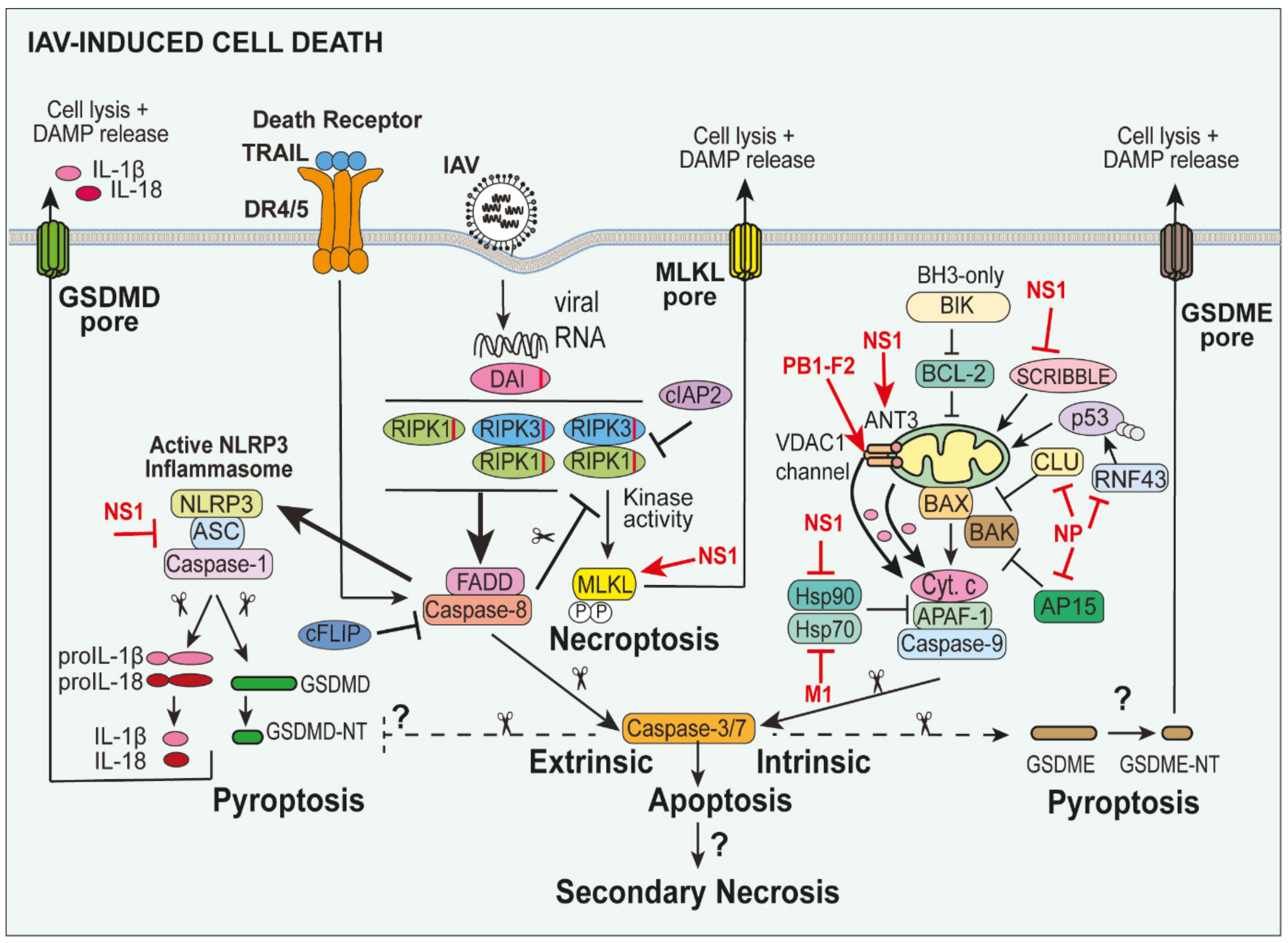
4. Cell Death Pathways and Their Activation during IAV Infection
5. NLRP3 Inflammasome-Associated Pyroptosis in IAV Infection
6. Apoptosis in IAV Infection
6.1. Extrinsic Apoptosis in IAV
6.2. Intrinsic “Mitochondrial-Dependent” Apoptosis in IAV Infection
6.3. Necroptosis Pathways in IAV
6.4. DAI-RIPK3-Driven Apoptotic Caspase-8 and Necroptotic MLKL during IAV Infection
7. Potential for Apoptotic Caspase-3-Induced Gasdermin E (GSDME)-Mediated Pyroptosis in IAV
8. Necrosis and Secondary Necrosis during IAV Infection
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, N.P.A.S.; Mueller, J. Updating the accounts: Global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull. Hist. Med. 2002, 76, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.-Y.; Zhou, B.; Wang, J.; Chai, Y.; Shen, Y.; Chen, X.; Ma, C.; Hong, W.; Chen, Y.; Zhang, Y.; et al. Dissemination, divergence and establishment of H7N9 influenza viruses in China. Nature 2015, 522, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Król, E.; Rychłowska, M.; Szewczyk, B. Antivirals—Current trends in fighting influenza. Acta Biochim. Pol. 2014, 61, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, N. Debate on whether Tamiflu prevents flu deaths reignites after new analysis. BMJ 2016, 353, i3077. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Denney, L.; Ho, L.-P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef]
- Tate, M.D.; Schilter, H.C.; Brooks, A.G.; Reading, P.C. Responses of Mouse Airway Epithelial Cells and Alveolar Macrophages to Virulent and Avirulent Strains of Influenza A Virus. Viral Immunol. 2011, 24, 77–88. [Google Scholar] [CrossRef]
- Tate, M.D.; Pickett, D.L.; van Rooijen, N.; Brooks, A.G.; Reading, P.C. Critical Role of Airway Macrophages in Modulating Disease Severity during Influenza Virus Infection of Mice. J. Virol. 2010, 84, 7569–7580. [Google Scholar] [CrossRef]
- Rodgers, B.; Mims, C.A. Interaction of influenza virus with mouse macrophages. Infect. Immun. 1981, 31, 751–757. [Google Scholar] [CrossRef]
- Thomas, B.J.; Porritt, R.A.; Hertzog, P.J.; Bardin, P.G.; Tate, M.D. Glucocorticosteroids enhance replication of respiratory viruses: Effect of adjuvant interferon. Sci. Rep. 2014, 4, 7176. [Google Scholar] [CrossRef]
- Tate, M.D.; Brooks, A.G.; Reading, P.C.; Mintern, J.D. Neutrophils sustain effective CD8+ T-cell responses in the respiratory tract following influenza infection. Immunol. Cell Biol. 2012, 90, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Gazit, R.; Gruda, R.; Elboim, M.; Arnon, T.I.; Katz, G.; Achdout, H.; Hanna, J.; Qimron, U.; Landau, G.; Greenbaum, E.; et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 2006, 7, 517–523. [Google Scholar] [CrossRef]
- Tate, M.D.; Ong, J.D.H.; Dowling, J.K.; McAuley, J.L.; Robertson, A.B.; Latz, E.; Drummond, G.R.; Cooper, M.A.; Hertzog, P.J.; Mansell, A. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci. Rep. 2016, 6, 27912. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.D.H.; Mansell, A.; Tate, M.D. Hero turned villain: NLRP3 inflammasome-induced inflammation during influenza A virus infection. J. Leukoc. Biol. 2017, 101, 863–874. [Google Scholar] [CrossRef]
- Michelle D, T.; Ashley, M. An update on the NLRP3 inflammasome and influenza: The road to redemption or perdition? Curr. Opin. Immunol. 2018, 54, 80–85. [Google Scholar] [CrossRef]
- Thomas, P.G.; Dash, P.; Aldridge, J.R.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.-Y. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef]
- Pinar, A.; Dowling, J.K.; Bitto, N.J.; Robertson, A.A.B.; Latz, E.; Stewart, C.R.; Drummond, G.R.; Cooper, M.A.; McAuley, J.L.; Tate, M.D.; et al. PB1-F2 Peptide Derived from Avian Influenza A Virus H7N9 Induces Inflammation via Activation of the NLRP3 Inflammasome. J. Biol. Chem. 2017, 292, 826–836. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Kuriakose, T.; Guy, C.S.; Samir, P.; Malireddi, R.K.S.; Mishra, A.; Kanneganti, T.-D. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 2017, 214, 2217–2229. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis E Sousa, C.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 2019, 97, 840–852. [Google Scholar] [CrossRef]
- Chung, W.-C.; Kang, H.-R.; Yoon, H.; Kang, S.-J.; Ting, J.P.-Y.; Song, M.J. Influenza A Virus NS1 Protein Inhibits the NLRP3 Inflammasome. PLoS ONE 2015, 10, e0126456. [Google Scholar] [CrossRef]
- Moriyama, M.; Chen, I.-Y.; Kawaguchi, A.; Koshiba, T.; Nagata, K.; Takeyama, H.; Hasegawa, H.; Ichinohe, T. The RNA- and TRIM25-Binding Domains of Influenza Virus NS1 Protein Are Essential for Suppression of NLRP3 Inflammasome-Mediated Interleukin-1β Secretion. J. Virol. 2016, 90, 4105–4114. [Google Scholar] [CrossRef]
- Park, H.-S.; Liu, G.; Thulasi Raman, S.N.; Landreth, S.L.; Liu, Q.; Zhou, Y. NS1 Protein of 2009 Pandemic Influenza A Virus Inhibits Porcine NLRP3 Inflammasome-Mediated Interleukin-1 Beta Production by Suppressing ASC Ubiquitination. J. Virol. 2018, 92, e00022-18. [Google Scholar] [CrossRef]
- Kandasamy, M.; Suryawanshi, A.; Tundup, S.; Perez, J.T.; Schmolke, M.; Manicassamy, S.; Manicassamy, B. RIG-I Signaling Is Critical for Efficient Polyfunctional T Cell Responses during Influenza Virus Infection. PLoS Pathog. 2016, 12, e1005754. [Google Scholar] [CrossRef]
- Wu, W.; Wang, X.; Zhang, W.; Tian, L.; Booth, J.L.; Duggan, E.S.; More, S.; Liu, L.; Dozmorov, M.; Metcalf, J.P. RIG-I Signaling via MAVS Is Dispensable for Survival in Lethal Influenza Infection In Vivo. Mediators Inflamm. 2018, 2018, 6808934. [Google Scholar] [CrossRef] [PubMed]
- Pang, I.K.; Pillai, P.S.; Iwasaki, A. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc. Natl. Acad. Sci. USA 2013, 110, 13910–13915. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hirohama, M.; Noguchi, M.; Nagata, K.; Kawaguchi, A. Influenza A Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J. Virol. 2018, 92, e00396-18. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.H.C.; Nicholls, J.M.; Ho, C.K.; Sia, S.F.; Mok, C.K.P.; Valkenburg, S.A.; Cheung, P.; Hui, K.P.Y.; Chan, R.W.Y.; Guan, Y.; et al. Highly pathogenic avian influenza A H5N1 and pandemic H1N1 virus infections have different phenotypes in Toll-like receptor 3 knockout mice. J. Gen. Virol. 2014, 95, 1870–1879. [Google Scholar] [CrossRef]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; Alexopoulou, L.; Escriou, N.; Flavell, R.; Chignard, M.; Si-Tahar, M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006, 2, e53. [Google Scholar] [CrossRef]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 Antagonist, Eritoran, Protects Mice from Lethal Influenza Infection. Nature 2013, 497, 498–502. [Google Scholar] [CrossRef]
- Shirey, K.A.; Lai, W.; Patel, M.C.; Pletneva, L.M.; Pang, C.; Kurt-Jones, E.; Lipsky, M.; Roger, T.; Calandra, T.; Tracey, K.; et al. Novel Strategies for Targeting Innate Immune Responses to Influenza. Mucosal Immunol. 2016, 9, 1173–1182. [Google Scholar] [CrossRef]
- Seo, S.-U.; Kwon, H.-J.; Song, J.-H.; Byun, Y.-H.; Seong, B.L.; Kawai, T.; Akira, S.; Kweon, M.-N. MyD88 Signaling Is Indispensable for Primary Influenza A Virus Infection but Dispensable for Secondary Infection. J. Virol. 2010, 84, 12713–12722. [Google Scholar] [CrossRef]
- Koyama, S.; Ishii, K.J.; Kumar, H.; Tanimoto, T.; Coban, C.; Uematsu, S.; Kawai, T.; Akira, S. Differential Role of TLR- and RLR-Signaling in the Immune Responses to Influenza A Virus Infection and Vaccination. J. Immunol. 2007, 179, 4711–4720. [Google Scholar] [CrossRef]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef]
- Rosli, S.; Kirby, F.J.; Lawlor, K.E.; Rainczuk, K.; Drummond, G.R.; Mansell, A.; Tate, M.D. Repurposing drugs targeting the P2X7 receptor to limit hyperinflammation and disease during influenza virus infection. Br. J. Pharmacol. 2019, 176, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, A.; Wan, Y.; Liu, X.; Qiu, C.; Xi, X.; Ren, Y.; Wang, J.; Dong, Y.; Bao, M.; et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 769–774. [Google Scholar] [CrossRef]
- Guo, J.; Huang, F.; Liu, J.; Chen, Y.; Wang, W.; Cao, B.; Zou, Z.; Liu, S.; Pan, J.; Bao, C.; et al. The Serum Profile of Hypercytokinemia Factors Identified in H7N9-Infected Patients can Predict Fatal Outcomes. Sci. Rep. 2015, 5, 10942. [Google Scholar] [CrossRef] [PubMed]
- Lauder, S.N.; Jones, E.; Smart, K.; Bloom, A.; Williams, A.S.; Hindley, J.P.; Ondondo, B.; Taylor, P.R.; Clement, M.; Fielding, C.; et al. Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur. J. Immunol. 2013, 43, 2613–2625. [Google Scholar] [CrossRef] [PubMed]
- Dienz, O.; Rud, J.G.; Eaton, S.M.; Lanthier, P.A.; Burg, E.; Drew, A.; Bunn, J.; Suratt, B.T.; Haynes, L.; Rincon, M. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012, 5, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, N.; Kurrer, M.; Bachmann, M.F.; Kopf, M. Interleukin-1 Is Responsible for Acute Lung Immunopathology but Increases Survival of Respiratory Influenza Virus Infection. J. Virol. 2005, 79, 6441–6448. [Google Scholar] [CrossRef]
- Liu, B.; Mori, I.; Hossain, M.J.; Dong, L.; Takeda, K.; Kimura, Y. Interleukin-18 improves the early defence system against influenza virus infection by augmenting natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2004, 85, 423–428. [Google Scholar] [CrossRef]
- Denton, A.E.; Doherty, P.C.; Turner, S.J.; La Gruta, N.L. IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8+ T cells. Eur. J. Immunol. 2007, 37, 368–375. [Google Scholar] [CrossRef]
- Seo, S.-U.; Kwon, H.-J.; Ko, H.-J.; Byun, Y.-H.; Seong, B.L.; Uematsu, S.; Akira, S.; Kweon, M.-N. Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog. 2011, 7, e1001304. [Google Scholar] [CrossRef]
- Lin, K.L.; Suzuki, Y.; Nakano, H.; Ramsburg, E.; Gunn, M.D. CCR2+ Monocyte-Derived Dendritic Cells and Exudate Macrophages Produce Influenza-Induced Pulmonary Immune Pathology and Mortality. J. Immunol. 2008, 180, 2562–2572. [Google Scholar] [CrossRef]
- Dawson, T.C.; Beck, M.A.; Kuziel, W.A.; Henderson, F.; Maeda, N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am. J. Pathol. 2000, 156, 1951–1959. [Google Scholar] [CrossRef]
- Lin, S.-J.; Lo, M.; Kuo, R.-L.; Shih, S.-R.; Ojcius, D.M.; Lu, J.; Lee, C.-K.; Chen, H.-C.; Lin, M.Y.; Leu, C.-M.; et al. The pathological effects of CCR2+ inflammatory monocytes are amplified by an IFNAR1-triggered chemokine feedback loop in highly pathogenic influenza infection. J. Biomed. Sci. 2014, 21, 99. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Steinmueller, M.; von Wulffen, W.; Cakarova, L.; Pinto, R.; Pleschka, S.; Mack, M.; Kuziel, W.A.; Corazza, N.; Brunner, T.; et al. Lung epithelial apoptosis in influenza virus pneumonia: The role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008, 205, 3065–3077. [Google Scholar] [CrossRef]
- Szretter, K.J.; Gangappa, S.; Belser, J.A.; Zeng, H.; Chen, H.; Matsuoka, Y.; Sambhara, S.; Swayne, D.E.; Tumpey, T.M.; Katz, J.M. Early control of H5N1 influenza virus replication by the type I interferon response in mice. J. Virol. 2009, 83, 5825–5834. [Google Scholar] [CrossRef]
- Szretter, K.J.; Gangappa, S.; Lu, X.; Smith, C.; Shieh, W.-J.; Zaki, S.R.; Sambhara, S.; Tumpey, T.M.; Katz, J.M. Role of Host Cytokine Responses in the Pathogenesis of Avian H5N1 Influenza Viruses in Mice. J. Virol. 2007, 81, 2736–2744. [Google Scholar] [CrossRef]
- Van Der Sluijs, K.F.; Van Elden, L.J.R.; Arens, R.; Nijhuis, M.; Schuurman, R.; Florquin, S.; Kwakkel, J.; Akira, S.; Jansen, H.M.; Lutter, R.; et al. Enhanced viral clearance in interleukin-18 gene-deficient mice after pulmonary infection with influenza A virus. Immunology 2005, 114, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.D.; Deng, Y.-M.; Jones, J.E.; Anderson, G.P.; Brooks, A.G.; Reading, P.C. Neutrophils Ameliorate Lung Injury and the Development of Severe Disease during Influenza Infection. J. Immunol. 2009, 183, 7441–7450. [Google Scholar] [CrossRef]
- Tate, M.D.; Ioannidis, L.J.; Croker, B.; Brown, L.E.; Brooks, A.G.; Reading, P.C. The role of neutrophils during mild and severe influenza virus infections of mice. PLoS ONE 2011, 6, e17618. [Google Scholar] [CrossRef]
- Nogusa, S.; Ritz, B.W.; Kassim, S.H.; Jennings, S.R.; Gardner, E.M. Characterization of age-related changes in natural killer cells during primary influenza infection in mice. Mech. Ageing Dev. 2008, 129, 223–230. [Google Scholar] [CrossRef]
- Hwang, I.; Scott, J.M.; Kakarla, T.; Duriancik, D.M.; Choi, S.; Cho, C.; Lee, T.; Park, H.; French, A.R.; Beli, E.; et al. Activation mechanisms of natural killer cells during influenza virus infection. PLoS ONE 2012, 7, e51858. [Google Scholar] [CrossRef]
- Taylor, R.M. Experimental infection with influenza a virus in mice: The increase in intrapulmonary virus after inoculation and the influence of various factors thereon. J. Exp. Med. 1941, 73, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.D.; Brooks, A.G.; Reading, P.C. Correlation between sialic acid expression and infection of murine macrophages by different strains of influenza virus. Microbes Infect. 2011, 13, 202–207. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A.; Durbin, R.K.; Zheng, H.; Palese, P.; Gertner, R.; Levy, D.E.; Durbin, J.E. The Role of Interferon in Influenza Virus Tissue Tropism. J. Virol. 1998, 72, 8550–8558. [Google Scholar] [CrossRef] [PubMed]
- Moisy, D.; Avilov, S.V.; Jacob, Y.; Laoide, B.M.; Ge, X.; Baudin, F.; Naffakh, N.; Jestin, J.-L. HMGB1 Protein Binds to Influenza Virus Nucleoprotein and Promotes Viral Replication. J. Virol. 2012, 86, 9122–9133. [Google Scholar] [CrossRef] [PubMed]
- Van den Brand, J.M.A.; Stittelaar, K.J.; van Amerongen, G.; Reperant, L.; de Waal, L.; Osterhaus, A.D.M.E.; Kuiken, T. Comparison of temporal and spatial dynamics of seasonal H3N2, pandemic H1N1 and highly pathogenic avian influenza H5N1 virus infections in ferrets. PLoS ONE 2012, 7, e42343. [Google Scholar] [CrossRef] [PubMed]
- Weinheimer, V.K.; Becher, A.; Tönnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Szymanski, K.; et al. Influenza A Viruses Target Type II Pneumocytes in the Human Lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Koo, G.-B.; Morgan, M.J.; Lee, D.-G.; Kim, W.-J.; Yoon, J.-H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Gaba, A.; Xu, F.; Lu, Y.; Park, H.-S.; Liu, G.; Zhou, Y. The NS1 Protein of Influenza A Virus Participates in Necroptosis by Interacting with MLKL and Increasing Its Oligomerization and Membrane Translocation. J. Virol. 2019, 93, e01835-18. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.-L.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.P.Y.; Li, H.S.; Cheung, M.C.; Chan, R.W.Y.; Yuen, K.M.; Mok, C.K.P.; Nicholls, J.M.; Peiris, J.S.M.; Chan, M.C.W. Highly pathogenic avian influenza H5N1 virus delays apoptotic responses via activation of STAT3. Sci. Rep. 2016, 6, 28593. [Google Scholar] [CrossRef] [PubMed]
- Van Krüchten, A.; Wilden, J.J.; Niemann, S.; Peters, G.; Löffler, B.; Ludwig, S.; Ehrhardt, C. Staphylococcus aureus triggers a shift from influenza virus–induced apoptosis to necrotic cell death. FASEB J. 2018, 32, 2779–2793. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, B.; Ghavami, S.; Rahim, M.N.; Klonisch, T.; Halayko, A.J.; Coombs, K.M. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim. Biophys. Acta BBA Mol. Cell Res. 2018, 1865, 364–378. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Tipper, J.; Chand, H.S.; Walton, S.; Harrod, K.S.; Tesfaigzi, Y. Bik Mediates Caspase-Dependent Cleavage of Viral Proteins to Promote Influenza A Virus Infection. Am. J. Respir. Cell Mol. Biol. 2016, 54, 664–673. [Google Scholar] [CrossRef]
- Arndt, U.; Wennemuth, G.; Barth, P.; Nain, M.; Al-Abed, Y.; Meinhardt, A.; Gemsa, D.; Bacher, M. Release of Macrophage Migration Inhibitory Factor and CXCL8/Interleukin-8 from Lung Epithelial Cells Rendered Necrotic by Influenza A Virus Infection. J. Virol. 2002, 76, 9298–9306. [Google Scholar] [CrossRef]
- Kosmider, B.; Messier, E.M.; Janssen, W.J.; Nahreini, P.; Wang, J.; Hartshorn, K.L.; Mason, R.J. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir. Res. 2012, 13, 43. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Morens, D.M. The Pathology of Influenza Virus Infections. Annu. Rev. Pathol. 2008, 3, 499–522. [Google Scholar] [CrossRef]
- Mauad, T.; Hajjar, L.A.; Callegari, G.D.; da Silva, L.F.F.; Schout, D.; Galas, F.R.B.G.; Alves, V.A.F.; Malheiros, D.M.A.C.; Auler, J.O.C.; Ferreira, A.F.; et al. Lung Pathology in Fatal Novel Human Influenza A (H1N1) Infection. Am. J. Respir. Crit. Care Med. 2010, 181, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, Q.; Florence, J.M.; Jung, B.-G.; Kurdowska, A.K.; Samten, B.; Idell, S.; Tang, H. Influenza Virus Infection Induces ZBP1 Expression and Necroptosis in Mouse Lungs. Front. Cell. Infect. Microbiol. 2019, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.Y.; Zhang, A.J.X.; Chu, H.; Li, C.; Zhu, H.; Mak, W.W.N.; Chen, Y.; Kok, K.-H.; To, K.K.W.; Yuen, K.-Y. H7N9 influenza A virus activation of necroptosis in human monocytes links innate and adaptive immune responses. Cell Death Dis. 2019, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Stasakova, J.; Ferko, B.; Kittel, C.; Sereinig, S.; Romanova, J.; Katinger, H.; Egorov, A. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1β and 18. J. Gen. Virol. 2005, 86, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.S.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.-D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef]
- Kuriakose, T.; Zheng, M.; Neale, G.; Kanneganti, T.-D. IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J. Immunol. 2018, 200, 1489–1495. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The pore forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Liu, F.; Zhang, X.; Liu, P.; Bajrami, B.; Teng, Y.; Zhao, L.; Zhou, S.; Yu, H.; Zhou, W.; et al. Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep. 2018, 22, 2924–2936. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8 dependent cleavage of Gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef]
- Takizawa, T.; Matsukawa, S.; Higuchi, Y.; Nakamura, S.; Nakanishi, Y.; Fukuda, R. Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. J. Gen. Virol. 1993, 74, 2347–2355. [Google Scholar] [CrossRef]
- Mori, I.; Komatsu, T.; Takeuchi, K.; Nakakuki, K.; Sudo, M.; Kimura, Y. In vivo induction of apoptosis by influenza virus. J. Gen. Virol. 1995, 76, 2869–2873. [Google Scholar] [CrossRef] [PubMed]
- Fesq, H.; Bacher, M.; Nain, M.; Gemsa, D. Programmed Cell Death (Apoptosis) in Human Monocytes Infected by Influenza A Virus. Immunobiology 1994, 190, 175–182. [Google Scholar] [CrossRef]
- Hofmann, P.; Sprenger, H.; Kaufmann, A.; Bender, A.; Hasse, C.; Nain, M.; Gemsa, D. Susceptibility of mononuclear phagocytes to influenza A virus infection and possible role in the antiviral response. J. Leukoc. Biol. 1997, 61, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Albert, M.; Reddy, A.; Feldman, M.; Sauter, B.; Kaplan, G.; Hellman, W.; Bhardwaj, N. The Distinctive Features of Influenza Virus Infection of Dendritic Cells. Immunobiology 1998, 198, 552–567. [Google Scholar] [CrossRef]
- Fernandez, M.V.; Miller, E.; Krammer, F.; Gopal, R.; Greenbaum, B.D.; Bhardwaj, N. Ion efflux and influenza infection trigger NLRP3 inflammasome signaling in human dendritic cells. J. Leukoc. Biol. 2016, 99, 723–734. [Google Scholar] [CrossRef]
- Thomas, J.M.; Pos, Z.; Reinboth, J.; Wang, R.Y.; Wang, E.; Frank, G.M.; Lusso, P.; Trinchieri, G.; Alter, H.J.; Marincola, F.M.; et al. Differential Responses of Plasmacytoid Dendritic Cells to Influenza Virus and Distinct Viral Pathogens. J. Virol. 2014, 88, 10758–10766. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mao, H.; Tu, W.; Qin, G.; Law, H.K.W.; Sia, S.F.; Chan, P.-L.; Liu, Y.; Lam, K.-T.; Zheng, J.; Peiris, M.; et al. Influenza Virus Directly Infects Human Natural Killer Cells and Induces Cell Apoptosis. J. Virol. 2009, 83, 9215–9222. [Google Scholar] [CrossRef]
- Mao, H.; Liu, Y.; Sia, S.F.; Peiris, J.S.M.; Lau, Y.-L.; Tu, W. Avian influenza virus directly infects human natural killer cells and inhibits cell activity. Virol. Sin. 2017, 32, 122–129. [Google Scholar] [CrossRef]
- Colamussi, M.L.; White, M.R.; Crouch, E.; Hartshorn, K.L. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood 1999, 93, 2395–2403. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, R.; Li, C.; Jiang, L.; Xiang, M.; Ye, Z.; Kita, H.; Melnick, A.M.; Dent, A.L.; Sun, J. BCL6 modulates tissue neutrophil survival and exacerbates pulmonary inflammation following influenza virus infection. Proc. Natl. Acad. Sci. USA 2019, 116, 11888–11893. [Google Scholar] [CrossRef]
- Kumar, R.; Herbert, P.E.; Warrens, A.N. An introduction to death receptors in apoptosis. Int. J. Surg. 2005, 3, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: Not so silently deadly. Clin. Transl. Immunol. 2017, 6, e124. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the Linear Ubiquitin Chain Assembly Complex Stabilizes the TNF-R1 Signaling Complex and Is Required for TNF-Mediated Gene Induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- Füllsack, S.; Rosenthal, A.; Wajant, H.; Siegmund, D. Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell Death Dis. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, J.; Zoueva, O.; Zhao, J.; Ye, Z.; Hewlett, I. Novel pandemic influenza A (H1N1) virus infection modulates apoptotic pathways that impact its replication in A549 cells. Microbes Infect. 2014, 16, 178–186. [Google Scholar] [CrossRef]
- Fujikura, D.; Chiba, S.; Muramatsu, D.; Kazumata, M.; Nakayama, Y.; Kawai, T.; Akira, S.; Kida, H.; Miyazaki, T. Type-I interferon is critical for FasL expression on lung cells to determine the severity of influenza. PLoS ONE 2013, 8, e55321. [Google Scholar] [CrossRef]
- Högner, K.; Wolff, T.; Pleschka, S.; Plog, S.; Gruber, A.D.; Kalinke, U.; Walmrath, H.-D.; Bodner, J.; Gattenlöhner, S.; Lewe-Schlosser, P.; et al. Macrophage-expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 2013, 9, e1003188. [Google Scholar] [CrossRef]
- Ishikawa, E.; Nakazawa, M.; Yoshinari, M.; Minami, M. Role of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Immune Response to Influenza Virus Infection in Mice. J. Virol. 2005, 79, 7658–7663. [Google Scholar] [CrossRef]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R.; Newmeyer, D.D. Bid, Bax, and Lipids Cooperate to Form Supramolecular Openings in the Outer Mitochondrial Membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Kim, H.-E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Timmer, J.; Sperandio, S.; Salvesen, G.S. The Apoptosome Activates Caspase-9 by Dimerization. Mol. Cell 2006, 22, 269–275. [Google Scholar] [CrossRef]
- Twiddy, D.; Cohen, G.M.; MacFarlane, M.; Cain, K. Caspase-7 Is Directly Activated by the ~700-kDa Apoptosome Complex and Is Released as a Stable XIAP-Caspase-7 ~200-kDa Complex. J. Biol. Chem. 2006, 281, 3876–3888. [Google Scholar] [CrossRef] [PubMed]
- Edison, N.; Zuri, D.; Maniv, I.; Bornstein, B.; Lev, T.; Gottfried, Y.; Kemeny, S.; Garcia-Fernandez, M.; Kagan, J.; Larisch, S. The IAP-antagonist ARTS initiates caspase activation upstream of cytochrome C and SMAC/Diablo. Cell Death Differ. 2012, 19, 356–368. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar] [CrossRef]
- McLean, J.E.; Datan, E.; Matassov, D.; Zakeri, Z.F. Lack of Bax Prevents Influenza A Virus-Induced Apoptosis and Causes Diminished Viral Replication. J. Virol. 2009, 83, 8233–8246. [Google Scholar] [CrossRef]
- Varga, Z.T.; Palese, P. The influenza A virus protein PB1-F2. Virulence 2011, 2, 542–546. [Google Scholar] [CrossRef]
- Bian, Q.; Lu, J.; Zhang, L.; Chi, Y.; Li, Y.; Guo, H. Highly pathogenic avian influenza A virus H5N1 non-structural protein 1 is associated with apoptotic activation of the intrinsic mitochondrial pathway. Exp. Ther. Med. 2017, 14, 4041–4046. [Google Scholar] [CrossRef]
- Yan, Y.; Du, Y.; Wang, G.; Deng, Y.; Li, R.; Li, K. The Novel H7N9 Influenza A Virus NS1 Induces p53-Mediated Apoptosis of A549 Cells. Cell. Physiol. Biochem. 2016, 38, 1447–1458. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Zhou, X.; Yang, Z.; Liu, X.; Cao, Z.; Song, H.; He, Y.; Huang, P. The NS1 protein of influenza A virus interacts with heat shock protein Hsp90 in human alveolar basal epithelial cells: Implication for virus-induced apoptosis. Virol. J. 2011, 8, 181. [Google Scholar] [CrossRef]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The ESEV PDZ-Binding Motif of the Avian Influenza A Virus NS1 Protein Protects Infected Cells from Apoptosis by Directly Targeting Scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar] [CrossRef]
- Tripathi, S.; Batra, J.; Cao, W.; Sharma, K.; Patel, J.R.; Ranjan, P.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; et al. Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: Implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis. 2013, 4, e562. [Google Scholar] [CrossRef]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef] [PubMed]
- Nailwal, H.; Sharma, S.; Mayank, A.K.; Lal, S.K. The nucleoprotein of influenza A virus induces p53 signaling and apoptosis via attenuation of host ubiquitin ligase RNF43. Cell Death Dis. 2015, 6, e1768. [Google Scholar] [CrossRef] [PubMed]
- Halder, U.C.; Bagchi, P.; Chattopadhyay, S.; Dutta, D.; Chawla-Sarkar, M. Cell death regulation during influenza A virus infection by matrix (M1) protein: A model of viral control over the cellular survival pathway. Cell Death Dis. 2011, 2, e197. [Google Scholar] [CrossRef] [PubMed]
- Kakkola, L.; Denisova, O.V.; Tynell, J.; Viiliäinen, J.; Ysenbaert, T.; Matos, R.C.; Nagaraj, A.; Ohman, T.; Kuivanen, S.; Paavilainen, H.; et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 2013, 4, e742. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef]
- Johnston, A.; Wang, Z. Necroptosis: MLKL Polymerization. J. Nat. Sci. 2018, 4, e513. [Google Scholar] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, B.M.; Albrecht, R.A.; Zaslavsky, E.; Nudelman, G.; Pincas, H.; Marjanovic, N.; Schotsaert, M.; Martínez-Romero, C.; Fenutria, R.; Ingram, J.P.; et al. Pandemic H1N1 influenza A viruses suppress immunogenic RIPK3-driven dendritic cell death. Nat. Commun. 2017, 8, 1931. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Sai, X.-Y.; Qian, X.-F.; Wu, Y.; Zou, L.-F.; Wang, H.-M.; Bian, T.; Yan, Z. Close Relationship between cIAP2 and Human ARDS Induced by Severe H7N9 Infection. BioMed Res. Int. 2019, 2019, 2121357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H.; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Downey, J.; Pernet, E.; Coulombe, F.; Allard, B.; Meunier, I.; Jaworska, J.; Qureshi, S.; Vinh, D.C.; Martin, J.G.; Joubert, P.; et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 2017, 13, e1006326. [Google Scholar] [CrossRef]
- Xu, Y.-L.; Tang, H.-L.; Peng, H.-R.; Zhao, P.; Qi, Z.-T.; Wang, W. RIP3 deficiency ameliorates inflammatory response in mice infected with influenza H7N9 virus infection. Oncotarget 2017, 8, 27715–27724. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Koehler, H.S.; Feng, Y.; Mandal, P.; Mocarski, E.S. Recognizing limits of Z-nucleic acid binding protein (ZBP1/DAI/DLM1) function. FEBS J. 2020. [Google Scholar] [CrossRef]
- Shaw, A.E.; Hughes, J.; Gu, Q.; Behdenna, A.; Singer, J.B.; Dennis, T.; Orton, R.J.; Varela, M.; Gifford, R.J.; Wilson, S.J.; et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 2017, 15, e2004086. [Google Scholar] [CrossRef]
- Neerukonda, N.S.; Katneni, U. Avian Pattern Recognition Receptor Sensing and Signaling. Vet. Sci. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef]
- Rodrigue-Gervais, I.G.; Labbé, K.; Dagenais, M.; Dupaul-Chicoine, J.; Champagne, C.; Morizot, A.; Skeldon, A.; Brincks, E.L.; Vidal, S.M.; Griffith, T.S.; et al. Cellular Inhibitor of Apoptosis Protein cIAP2 Protects against Pulmonary Tissue Necrosis during Influenza Virus Infection to Promote Host Survival. Cell Host Microbe 2014, 15, 23–35. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Feltham, R.; Yabal, M.; Conos, S.A.; Chen, K.W.; Ziehe, S.; Graß, C.; Zhan, Y.; Nguyen, T.A.; Hall, C.; et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1β Activation and Cell Death as a Consequence of TLR-MyD88-Induced cIAP1-TRAF2 Degradation. Cell Rep. 2017, 20, 668–682. [Google Scholar] [CrossRef]
- Yabal, M.; Müller, N.; Adler, H.; Knies, N.; Groß, C.J.; Damgaard, R.B.; Kanegane, H.; Ringelhan, M.; Kaufmann, T.; Heikenwälder, M.; et al. XIAP Restricts TNF- and RIP3-Dependent Cell Death and Inflammasome Activation. Cell Rep. 2014, 7, 1796–1808. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Death Pathway | Extrinsic Apoptosis | Intrinsic Apoptosis | GSDMD-Mediated Pyroptosis | GSDME-Mediated Pyroptosis | Necroptosis |
|---|---|---|---|---|---|
| Regulation | Regulated | Regulated | Regulated | Regulated | Regulated |
| Activators | Death receptor and ZBP1 ligation. | IAV PB1-F2, DNA damage and metabolic stress. | ATP, IAV viral RNA, IAV Matrix 2 and PB1-F2 proteins. | Not determined. | Death receptors and TLR ligation. |
| Morphological features | Plasma membrane blebbing, cell shrinkage, apoptotic bodies, nuclear and DNA fragmentation. | Plasma membrane blebbing, cell shrinkage, apoptotic bodies, nuclear and DNA fragmentation. | Disruption of plasma membrane, cell swelling. | Disruption of plasma membrane, cell swelling. | Disruption of plasma membrane and cell swelling. |
| Mediating Effectors | RIPK1, RIPK3, FADD and Caspase-8, -3 and -7. | BAX, BAK, APAF-1, cytochrome c and Caspase-9, -3 and -7. | NLRP3, Caspase-1, -4, -5, -8, -11 and GSDMD. | Caspase-3 and GSDME. | RIPK1, RIPK3 and MLKL. |
| Host Inhibitory Molecules | cFLIP, cIAP1/2 and XIAP. | BCL-2 pro-survival family e.g., BCL-2, BCL-XL and XIAP. | C-terminal domain of GSDMD and Caspase 3. | C-terminal domain of GSDME. | cIAP1/2 and Caspase-8. |
| Key Biochemical Readout | Caspase-8 cleavage. | Caspase-9 cleavage. | GSDMD cleavage. | GSDME cleavage. | MLKL phosphorylation. |
| Release of cellular contents | No | No | Yes | Yes | Yes |
| Inflammatory | −/+ | −/+ | +++ | ++ | ++ |
| Cell Type | Extrinsic Apoptosis | Intrinsic Apoptosis | Necrosis (Primary or Secondary?) | Necroptosis | Pyroptosis |
|---|---|---|---|---|---|
| Airway Epithelial cells | Human primary type I-like alveolar epithelial cells [74]. | Human primary type I-like alveolar epithelial cells [74]. | Human primary bronchial epithelial cells [78]. | Murine primary type I alveolar epithelial cells [82]. | Human primary bronchial epithelial cells [33]. |
| Human lung adenocarcinoma A549 type II cell line [75]. | Human lung adenocarcinoma A549 type II cell line [75,76]. | Human primary alveolar type I-like and type II epithelial cells [79]. | Murine immortalised LET1 type I alveolar epithelial cells [23]. | ||
| Murine immortalised LET1 type I alveolar epithelial cells [23]. | Murine primary tracheal epithelial cells [77]. | Epithelial cells in human lung tissue sections [80,81]. | |||
| Macrophages and monocytes | Human blood-derived monocytes [83]. | Human blood-derived monocytes [83]. | Human blood-derived monocytes [83]. | Human monocyte-derived macrophages [84]. | |
| Human monocyte-derived macrophages [84]. | Murine primary bone-marrow derived macrophages [82,86]. | ||||
| Murine primary bone-marrow derived macrophages [24,85,86]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laghlali, G.; Lawlor, K.E.; Tate, M.D. Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death. Viruses 2020, 12, 401. https://doi.org/10.3390/v12040401
Laghlali G, Lawlor KE, Tate MD. Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death. Viruses. 2020; 12(4):401. https://doi.org/10.3390/v12040401
Chicago/Turabian StyleLaghlali, Gabriel, Kate E. Lawlor, and Michelle D. Tate. 2020. "Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death" Viruses 12, no. 4: 401. https://doi.org/10.3390/v12040401
APA StyleLaghlali, G., Lawlor, K. E., & Tate, M. D. (2020). Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death. Viruses, 12(4), 401. https://doi.org/10.3390/v12040401