Host–Virus Interaction: How Host Cells Defend against Influenza A Virus Infection



{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. IAV Viral Proteins
1.2. Antigenic Shift and Antigenic Drift
2. Influenza A Virus Host Adaption and Life Cycle
2.1. Influenza A Virus Host Switch Events
2.2. Life Cycle of Influenza A Virus
3. Host Immune Responses Against IAV Infection
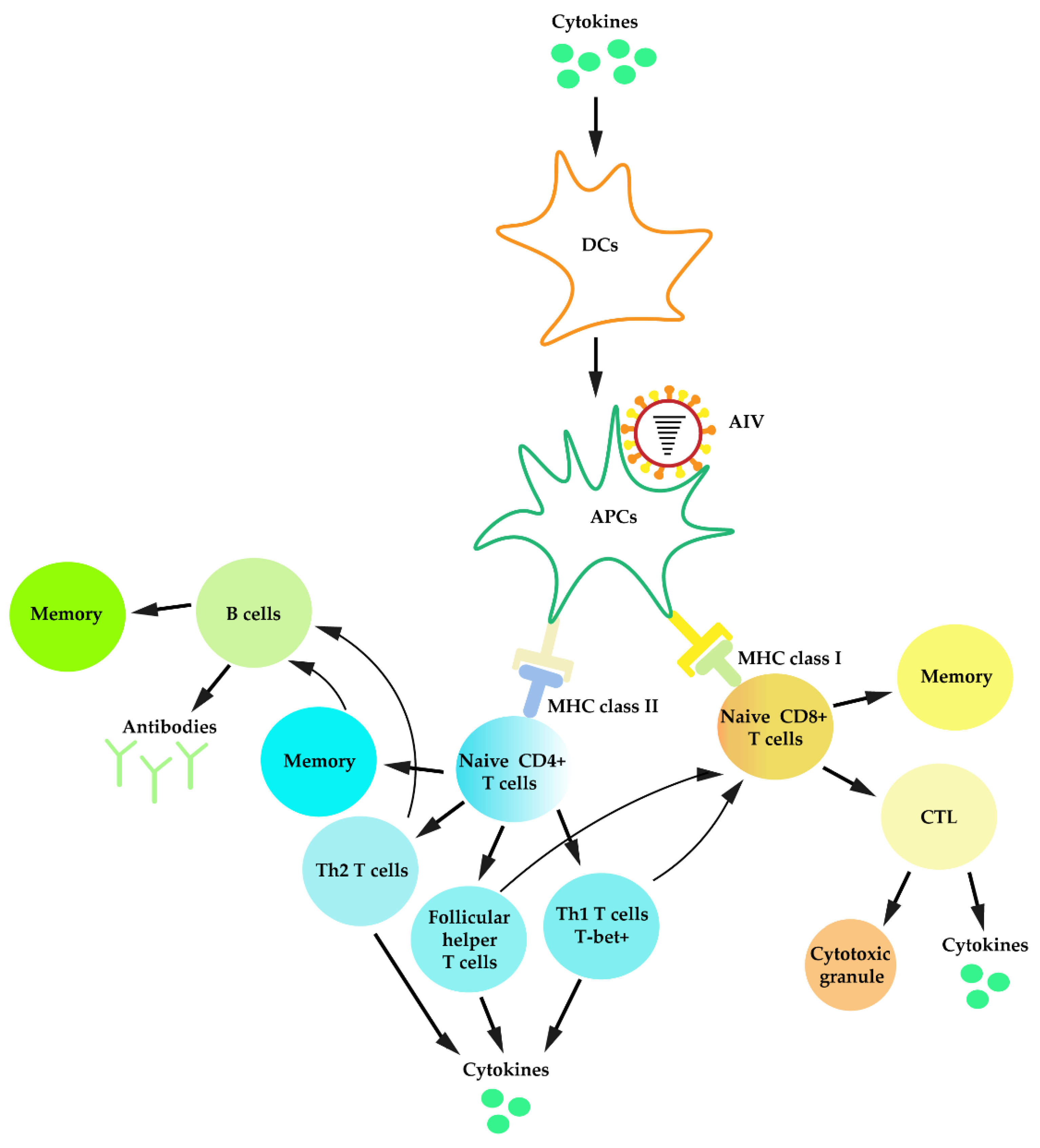
3.1. Immune Cells Involved in IAV Infection
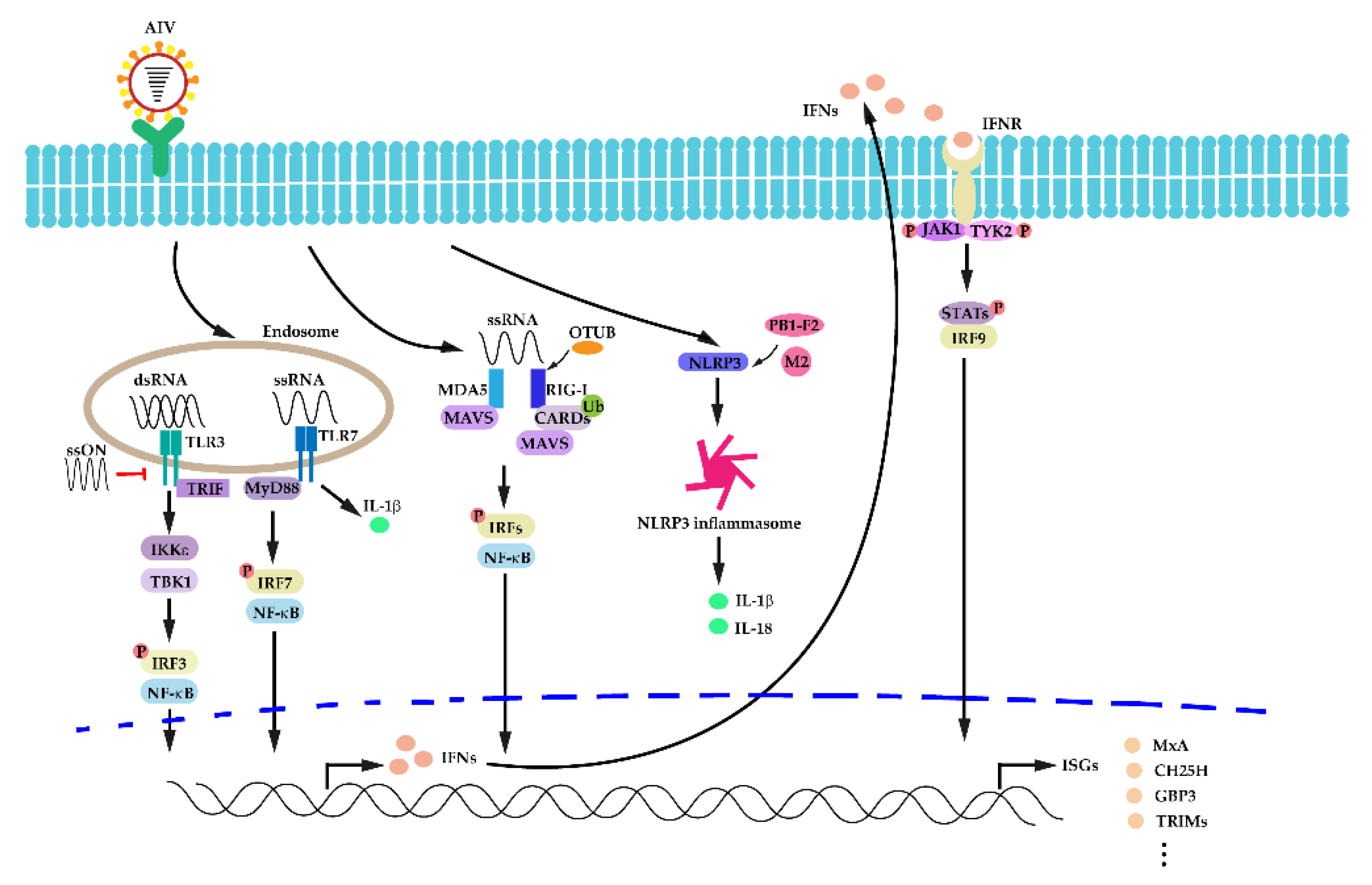
3.2. Activation of Innate Immunity in IAV Infection
3.3. The Host Interferon (IFN) Response in IAV Infection
3.4. Autophagy
3.5. Adaptive Immunity against IAV Infection
3.6. Apoptosis against IAV Infection
3.6.1. The Intrinsic and Extrinsic Apoptosis Pathway
3.6.2. Apoptosis after IAV Infection
4. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nikitin, N.; Petrova, E.D.; Trifonova, E.; Karpova, O. Influenza Virus Aerosols in the Air and Their Infectiousness. Adv. Virol. 2014, 2014, 859090. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. Influenza: The once and future pandemic. Public Health Rep. 2010, 125, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Watanabe, M.; Goto, H.; Kawaoka, Y. Identification of a Novel Viral Protein Expressed from the PB2 Segment of Influenza A Virus. J. Virol. 2015, 90, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.-A.; Wills, N.M.; Xiao, Y.-L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef]
- Muramoto, Y.; Noda, T.; Kawakami, E.; Akkina, R.; Kawaoka, Y. Identification of Novel Influenza A Virus Proteins Translated from PA mRNA. J. Virol. 2012, 87, 2455–2462. [Google Scholar] [CrossRef]
- Wise, H.M.; Hutchinson, E.C.; Jagger, B.W.; Stuart, A.D.; Kang, Z.H.; Robb, N.; Schwartzman, L.M.; Kash, J.C.; Fodor, E.; Firth, A.E.; et al. Identification of a Novel Splice Variant Form of the Influenza A Virus M2 Ion Channel with an Antigenically Distinct Ectodomain. PLoS Pathog. 2012, 8, e1002998. [Google Scholar] [CrossRef]
- Selman, M.; Dankar, S.K.; Forbes, N.; Jia, J.-J.; Brown, E.G. Adaptive mutation in influenza A virus non-structural gene is linked to host switching and induces a novel protein by alternative splicing. Emerg. Microbes Infect. 2012, 1, e42. [Google Scholar] [CrossRef]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.A.; Chen, L.-M.; Recuenco-Cabrera, S.; Ellison, J.A.; Davis, C.T.; York, I.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco-Cabrera, S.; Gómez, J.; et al. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Su, S.; Gu, M.; Liu, D.; Cui, J.; Gao, G.F.; Zhou, J.; Liu, X. Epidemiology, Evolution, and Pathogenesis of H7N9 Influenza Viruses in Five Epidemic Waves since 2013 in China. Trends Microbiol. 2017, 25, 713–728. [Google Scholar] [CrossRef]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.B.; Van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Boyoglu-Barnum, S.; Hutchinson, G.B.; Boyington, J.C.; Moin, S.M.; Gillespie, R.A.; Tsybovsky, Y.; Stephens, T.; Vaile, J.R.; Lederhofer, J.; Corbett, K.S.; et al. Glycan repositioning of influenza hemagglutinin stem facilitates the elicitation of protective cross-group antibody responses. Nat. Commun. 2020, 11, 791. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gambaryan, A.; Klimov, A.; Castrucci, M.R.; Donatelli, I.; Kawaoka, Y. Early Alterations of the Receptor-Binding Properties of H1, H2, and H3 Avian Influenza Virus Hemagglutinins after Their Introduction into Mammals. J. Virol. 2000, 74, 8502–8512. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, S.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef] [PubMed]
- Shelton, H.; Ayora-Talavera, G.; Ren, J.; Loureiro, S.; Pickles, R.J.; Barclay, W.S.; Jones, I. Receptor binding profiles of avian influenza virus hemagglutinin subtypes on human cells as a predictor of pandemic potential. J. Virol. 2010, 85, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, M.; Fouchier, R.A. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 2014, 33, 823–841. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, W.; Wang, F.; Qi, J.; Wu, Y.; Song, H.; Gao, F.; Bi, Y.; Zhang, Y.; Fan, Z.; et al. Structures and Receptor Binding of Hemagglutinins from Human-Infecting H7N9 Influenza Viruses. Science 2013, 342, 243–247. [Google Scholar] [CrossRef]
- Xu, Y.; Peng, R.; Zhang, W.; Qi, J.; Song, H.; Liu, S.; Wang, H.; Wang, M.; Xiao, H.; Fu, L.; et al. Avian-to-Human Receptor-Binding Adaptation of Avian H7N9 Influenza Virus Hemagglutinin. Cell Rep. 2019, 29, 2217–2228. [Google Scholar] [CrossRef]
- Cohen, M.; Zhang, X.-Q.; Senaati, H.P.; Chen, H.; Varki, N.M.; Schooley, R.T.; Gagneux, P. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol. J. 2013, 10, 321. [Google Scholar] [CrossRef]
- Barnard, K.N.; Alford-Lawrence, B.K.; Buchholz, D.W.; Wasik, B.R.; LaClair, J.R.; Yu, H.; Honce, R.; Ruhl, S.; Pajic, P.; Daugherity, E.K.; et al. Modified sialic acids on mucus and erythrocytes inhibit influenza A HA and NA functions. J. Virol. 2020. [Google Scholar] [CrossRef]
- Air, G.M. Influenza neuraminidase. Influenza Other Respir. Viruses 2012, 6, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Evseenko, V.A.; Svyatchenko, S.V.; Kolosova, N.P.; Kovrizhkina, V.L.; Marchenko, V.Y.; Durymanov, A.G.; Goncharova, N.I.; Ryzhikov, A.B. Comparative thermostability analysis of zoonotic and human influenza virus A and B neuraminidase. Arch. Virol. 2019, 165, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Eichelberger, M.C.; Morens, D.M.; Taubenberger, J.K. Neuraminidase as an influenza vaccine antigen: A low hanging fruit, ready for picking to improve vaccine effectiveness. Curr. Opin. Immunol. 2018, 53, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Turner, H.L.; Lang, S.; McBride, R.; Bangaru, S.; Gilchuk, I.M.; Yu, W.; Paulson, J.C.; Crowe, J.E.; Ward, A.B.; et al. Structural Basis of Protection against H7N9 Influenza Virus by Human Anti-N9 Neuraminidase Antibodies. Cell Host Microbe 2019, 26, 729–738. [Google Scholar] [CrossRef]
- Noda, T.; Kawaoka, Y. Structure of influenza virus ribonucleoprotein complexes and their packaging into virions. Rev. Med. Virol. 2010, 20, 380–391. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Genet. 2015, 13, 28–41. [Google Scholar] [CrossRef]
- Velthuis, A.T.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Genet. 2016, 14, 479–493. [Google Scholar] [CrossRef]
- König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.-H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, N.M.; Ortigoza, M.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.-P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef]
- Yang, C.; Liu, X.; Gao, Q.; Cheng, T.; Xiao, R.; Ming, F.; Zhang, S.; Jin, M.; Chen, H.; Ma, W.; et al. The Nucleolar Protein LYAR Facilitates Ribonucleoprotein Assembly of Influenza A Virus. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, W.; Chen, Q.; Huang, W.; Yang, Z.; Li, X.; Wang, X.-H. Inhibition viral RNP and anti-inflammatory activity of coumarins against influenza virus. Biomed. Pharmacother. 2017, 87, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Lai, C.J.; Choppin, P.W. Sequences of mRNAs derived from genome RNA segment 7 of influenza virus: Colinear and interrupted mRNAs code for overlapping proteins. Proc. Natl. Acad. Sci. USA 1981, 78, 4170–4174. [Google Scholar] [CrossRef] [PubMed]
- Safo, M.K.; Musayev, F.N.; Mosier, P.D.; Zhou, Q.; Xie, H.; Desai, U.R. Crystal Structures of Influenza A Virus Matrix Protein M1: Variations on a Theme. PLoS ONE 2014, 9, e109510. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Lamb, R.A. The M2 Proton Channels of Influenza A and B Viruses. J. Biol. Chem. 2006, 281, 8997–9000. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Pekosz, A.; Shuck, K.; Pinto, L.H.; A Lamb, R. Influenza A Virus M2 Ion Channel Activity Is Essential for Efficient Replication in Tissue Culture. J. Virol. 2002, 76, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; Chakravarty, C.; Sellathamby, S.; Lal, S.K. The influenza A virus matrix protein 2 undergoes retrograde transport from the endoplasmic reticulum into the cytoplasm and bypasses cytoplasmic proteasomal degradation. Arch. Virol. 2016, 162, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Caplen, F.V.; Krug, R.M. Regulation of the extent of splicing of influenza virus NS1 mRNA: Role of the rates of splicing and of the nucleocytoplasmic transport of NS1 mRNA. Mol. Cell. Biol. 1991, 11, 1092–1098. [Google Scholar] [CrossRef]
- Skórko, R.; Summers, D.F.; Galarza, J.M. Influenza A virus in vitro transcription: Roles of NS1 and NP proteins in regulating RNA synthesis. Virology 1991, 180, 668–677. [Google Scholar] [CrossRef]
- Marc, D. Influenza virus non-structural protein NS1: Interferon antagonism and beyond. J. Gen. Virol. 2014, 95, 2594–2611. [Google Scholar] [CrossRef]
- Hale, B.; Randall, R.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Wilkins, C.; Gale, M. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; Schulz, O.; Pichlmair, A.; Bier, K.; Robb, N.; Vreede, F.; Barclay, W.S.; Fodor, E.; et al. RIG-I Detects Viral Genomic RNA during Negative-Strand RNA Virus Infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Xie, Y.; Muñoz-Moreno, R.; Wang, J.; Zhang, L.; Esparza, M.; García-Sastre, A.; Fontoura, B.M.A.; Ren, Y. Structural basis for influenza virus NS1 protein block of mRNA nuclear export. Nat. Microbiol. 2019, 4, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Baranovskaya, I.; Sergeeva, M.; Fadeev, A.; Kadirova, R.; Ivanova, A.; Ramsay, E.; Vasin, A. Changes in RNA secondary structure affect NS1 protein expression during early stage influenza virus infection. Virol. J. 2019, 16, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Han, C.W.; Jeong, M.S.; Jang, S.B. Structure and Function of the Influenza A Virus Non-Structural Protein 1. J. Microbiol. Biotechnol. 2019, 29, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Talon, J.; Palese, P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.; Simpson-Holley, M.; Archer, K.; Medcalf, L.; Hallam, R.; McCauley, J.; Digard, P. Interaction of the Influenza Virus Nucleoprotein with the Cellular CRM1-Mediated Nuclear Export Pathway. J. Virol. 2001, 75, 408–419. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Chen, Q.; Wang, H.; Yao, Y.; Chen, J.; Chen, Z. A Second CRM1-Dependent Nuclear Export Signal in the Influenza A Virus NS2 Protein Contributes to the Nuclear Export of Viral Ribonucleoproteins. J. Virol. 2012, 87, 767–778. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, X.; Zhang, A.; Zhou, H.; Liu, Z.; Chen, H.; Jin, M. CHD3 facilitates vRNP nuclear export by interacting with NES1 of influenza A virus NS2. Cell. Mol. Life Sci. 2014, 72, 971–982. [Google Scholar] [CrossRef]
- Zeng, L.-Y.; Yang, J.; Liu, S. Investigational hemagglutinin-targeted influenza virus inhibitors. Expert Opin. Investig. Drugs 2016, 26, 63–73. [Google Scholar] [CrossRef]
- Bui, C.; Chughtai, A.A.; Adam, D.C.; MacIntyre, C.R. An overview of the epidemiology and emergence of influenza A infection in humans over time. Arch. Public Health 2017, 75, 15. [Google Scholar] [CrossRef] [PubMed]
- Carrat, F.; Flahault, A. Influenza vaccine: The challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Madan, R.; Karp, C.; Braciale, T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Ciminski, K.; Thamamongood, T.; Zimmer, G.; Schwemmle, M. Novel insights into bat influenza A viruses. J. Gen. Virol. 2017, 98, 2393–2400. [Google Scholar] [CrossRef]
- Butt, K.M.; Smith, G.J.; Chen, H.; Zhang, L.J.; Leung, Y.H.C.; Xu, K.M.; Lim, W.; Webster, R.G.; Yuen, K.-Y.; Peiris, J.S.M.; et al. Human Infection with an Avian H9N2 Influenza A Virus in Hong Kong in 2003. J. Clin. Microbiol. 2005, 43, 5760–5767. [Google Scholar] [CrossRef]
- Chen, H.; Yuan, H.; Gao, R.; Zhang, J.; Wang, D.-Y.; Xiong, Y.; Fan, G.; Yang, F.; Li, X.; Zhou, J.; et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: A descriptive study. Lancet 2014, 383, 714–721. [Google Scholar] [CrossRef]
- Yang, Z.-F.; Mok, C.K.P.; Peiris, J.S.M.; Zhong, N. Human Infection with a Novel Avian Influenza A (H5N6) Virus. N. Engl. J. Med. 2015, 373, 487–489. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.-Y.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef]
- Shi, Y.; Wu, Y.; Zhang, W.; Qi, J.; Gao, G.F. Enabling the ’host jump’: Structural determinants of receptor-binding specificity in influenza A viruses. Nat. Rev. Genet. 2014, 12, 822–831. [Google Scholar] [CrossRef]
- De Vries, R.P.; Peng, W.; Grant, O.; Thompson, A.J.; Zhu, X.; Bouwman, K.M.; De La Pena, A.T.T.; Van Breemen, M.J.; Wickramasinghe, I.N.A.; De Haan, C.A.M.; et al. Three mutations switch H7N9 influenza to human-type receptor specificity. PLoS Pathog. 2017, 13, e1006390. [Google Scholar] [CrossRef]
- Stevens, J.; Blixt, O.; Tumpey, T.M.; Taubenberger, J.K.; Paulson, J.C.; Wilson, I.A. Structure and Receptor Specificity of the Hemagglutinin from an H5N1 Influenza Virus. Science 2006, 312, 404–410. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.; Du, W.; Guo, H.; De Haan, C.A. Influenza A Virus Hemagglutinin-Neuraminidase-Receptor Balance: Preserving Virus Motility. Trends Microbiol. 2020, 28, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Mistry, B.; Haslam, S.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Genet. 2019, 17, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.H. History and Epidemiology of Swine Influenza in Europe. Curr. Top. Microbiol. Immunol. 2011, 370, 133–146. [Google Scholar]
- Skehel, J.J.; Wiley, D.C. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.J.; Dhanasekaran, V.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Lee, B.E.; Patel, J.; Bastien, N.; Grimsrud, K.; Seal, R.F.; King, R.; Marshall, F.; Li, Y. Swine Influenza (H3N2) Infection in a Child and Possible Community Transmission, Canada. Emerg. Infect. Dis. 2007, 13, 1865–1870. [Google Scholar] [CrossRef]
- Herfst, S.; Imai, M.; Kawaoka, Y.; Fouchier, R. Avian Influenza Virus Transmission to Mammals. Curr. Top. Microbiol. Immunol. 2014, 385, 137–155. [Google Scholar]
- Iwasaki, Y.; Abe, T.; Wada, Y.; Wada, K.; Ikemura, T. Novel bioinformatics strategies for prediction of directional sequence changes in influenza virus genomes and for surveillance of potentially hazardous strains. BMC Infect. Dis. 2013, 13, 386. [Google Scholar] [CrossRef]
- Eng, C.L.P.; Tong, J.C.; Tan, T.W. Distinct Host Tropism Protein Signatures to Identify Possible Zoonotic Influenza A Viruses. PLoS ONE 2016, 11, e0150173. [Google Scholar] [CrossRef]
- Qiang, X.; Kou, Z.; Fang, G.; Wang, Y. Scoring Amino Acid Mutations to Predict Avian-to-Human Transmission of Avian Influenza Viruses. Molecules 2018, 23, 1584. [Google Scholar] [CrossRef] [PubMed]
- Di Giallonardo, F.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Li, B.; Hu, Y.; Kang, X.-P.; Wu, X.-Y.; Huang, M.-T.; Li, Y.-C.; Zhao, Z.-P.; Qin, C.-F.; et al. Machine Learning Methods for Predicting Human-Adaptive Influenza A Viruses Based on Viral Nucleotide Compositions. Mol. Biol. Evol. 2019, 37, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Babayan, S.A.; Orton, R.J.; Streicker, D.G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 2018, 362, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Weinheimer, V.K.; Becher, A.; Tönnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Szymanski, K.; et al. Influenza A Viruses Target Type II Pneumocytes in the Human Lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef]
- Müller, K.H.; Kakkola, L.; Nagaraj, A.S.; Cheltsov, A.V.; Anastasina, M.; Kainov, D. Emerging cellular targets for influenza antiviral agents. Trends Pharmacol. Sci. 2012, 33, 89–99. [Google Scholar] [CrossRef]
- Van Riel, D.; Bakker, M.A.D.; Leijten, L.M.; Chutinimitkul, S.; Munster, V.; De Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.; Osterhaus, A.D.; Kuiken, T. Seasonal and Pandemic Human Influenza Viruses Attach Better to Human Upper Respiratory Tract Epithelium than Avian Influenza Viruses. Am. J. Pathol. 2010, 176, 1614–1618. [Google Scholar] [CrossRef]
- Van Riel, D.; Munster, V.; De Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.; Osterhaus, A.D.; Kuiken, T. Human and Avian Influenza Viruses Target Different Cells in the Lower Respiratory Tract of Humans and Other Mammals. Am. J. Pathol. 2007, 171, 1215–1223. [Google Scholar] [CrossRef]
- Olofsson, S.; Kumlin, U.; Dimock, K.; Arnberg, N. Avian influenza and sialic acid receptors: More than meets the eye? Lancet Infect. Dis. 2005, 5, 184–188. [Google Scholar] [CrossRef]
- De Vries, E.; De Vries, R.P.; Wienholts, M.J.; Floris, C.E.; Jacobs, M.-S.; Heuvel, A.V.D.; Rottier, P.J.M.; De Haan, C.A.M. Influenza A virus entry into cells lacking sialylated N-glycans. Proc. Natl. Acad. Sci. USA 2012, 109, 7457–7462. [Google Scholar] [CrossRef]
- Gill, J.S.; Webby, R.; Gilchrist, M.J.; Gray, G.C. Avian influenza among waterfowl hunters and wildlife professionals. Emerg. Infect. Dis. 2006, 12, 1284–1286. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.W.; Mao, H.J.; Wu, Y.-L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.S.; Gludish, D.W.J.; Whittaker, G.R. Cleavage Activation of the Human-Adapted Influenza Virus Subtypes by Matriptase Reveals both Subtype and Strain Specificities. J. Virol. 2012, 86, 10579–10586. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-I.; Kim, Y.-I.; Park, S.-J.; Kim, E.-H.; Kim, S.; Si, Y.-J.; Song, M.-S.; Pascua, P.N.Q.; Govorkova, E.A.; Webster, R.G.; et al. A Novel Neuraminidase-Dependent Hemagglutinin Cleavage Mechanism Enables the Systemic Spread of an H7N6 Avian Influenza Virus. mBio 2019, 10, 10. [Google Scholar] [CrossRef]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Tsegaye, T.S.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; et al. Proteolytic Activation of the 1918 Influenza Virus Hemagglutinin. J. Virol. 2009, 83, 3200–3211. [Google Scholar] [CrossRef]
- Dietze, K.; Graaf, A.; Homeier-Bachmann, T.; Grund, C.; Forth, L.; Pohlmann, A.; Jeske, C.; Wintermann, M.; Beer, M.; Conraths, F.J.; et al. From low to high pathogenicity-Characterization of H7N7 avian influenza viruses in two epidemiologically linked outbreaks. Transbound. Emerg. Dis. 2018, 65, 1576–1587. [Google Scholar] [CrossRef]
- Gultyaev, A.P.; Spronken, M.I.; Richard, M.; Schrauwen, E.J.A.; Olsthoorn, R.; Fouchier, R. Subtype-specific structural constraints in the evolution of influenza A virus hemagglutinin genes. Sci. Rep. 2016, 6, 38892. [Google Scholar] [CrossRef]
- Boulo, S.; Akarsu, H.; Ruigrok, R.W.; Baudin, F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007, 124, 12–21. [Google Scholar] [CrossRef]
- Dias, A.; Bouvier, D.; Crepin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef]
- Rodriguez, P.; Marcos-Villar, L.; Zamarreño, N.; Yángüez, E.; Nieto, A. Mutations of the segment-specific nucleotides at the 3′ end of influenza virus NS segment control viral replication. Virology 2020, 539, 104–113. [Google Scholar] [CrossRef]
- Cheng, J.; Tao, J.; Li, B.; Shi, Y.; Liu, H. The tyrosine 73 and serine 83 dephosphorylation of H1N1 swine influenza virus NS1 protein attenuates virus replication and induces high levels of beta interferon. Virol. J. 2019, 16, 152. [Google Scholar] [CrossRef] [PubMed]
- Akarsu, H.; Burmeister, W.; Petosa, C.; Petit, I.; Müller, C.W.; Ruigrok, R.W.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jayaraman, A.; Maniprasad, P.; Raman, R.; Houser, K.V.; Pappas, C.; Zeng, H.; Sasisekharan, R.; Katz, J.M.; Tumpey, T.M. N-Linked Glycosylation of the Hemagglutinin Protein Influences Virulence and Antigenicity of the 1918 Pandemic and Seasonal H1N1 Influenza A Viruses. J. Virol. 2013, 87, 8756–8766. [Google Scholar] [CrossRef] [PubMed]
- Brett, K.; Kordyukova, L.V.; Serebryakova, M.V.; Mintaev, R.R.; Alexeevski, A.V.; Veit, M. Site-specific S-acylation of influenza virus hemagglutinin: the location of the acylation site relative to the membrane border is the decisive factor for attachment of stearate. J. Biol. Chem. 2014, 289, 34978–34989. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, T.; Momose, F.; Ichikawa, R.; Takeuchi, K.; Morikawa, Y. Influenza A Virus Hemagglutinin and Neuraminidase Mutually Accelerate Their Apical Targeting through Clustering of Lipid Rafts. J. Virol. 2014, 88, 10039–10055. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Okura, T.; Kawahara, M.; Takizawa, N.; Momose, F.; Morikawa, Y. Apical Trafficking Pathways of Influenza A Virus HA and NA via Rab17- and Rab23-Positive Compartments. Front. Microbiol. 2019, 10, 1857. [Google Scholar] [CrossRef] [PubMed]
- Amorim, M.J.; A Bruce, E.; Read, E.K.C.; Foeglein, Á.; Mahen, R.; Stuart, A.D.; Digard, P. A Rab11- and Microtubule-Dependent Mechanism for Cytoplasmic Transport of Influenza A Virus Viral RNA. J. Virol. 2011, 85, 4143–4156. [Google Scholar] [CrossRef]
- Rossman, J.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef]
- Palese, P.; Tobita, K.; Ueda, M.; Compans, R. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 1974, 61, 397–410. [Google Scholar] [CrossRef]
- Stertz, S.; Shaw, M. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011, 13, 516–525. [Google Scholar] [CrossRef]
- Tran, A.T.; Rahim, M.N.; Ranadheera, C.; Kroeker, A.; Cortens, J.P.; Opanubi, K.J.; A Wilkins, J.; Coombs, K.M. Knockdown of specific host factors protects against influenza virus-induced cell death. Cell Death Dis. 2013, 4, e769. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; Tenoever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Clohisey, S.M.; Chia, B.S.; Wang, B.; Cui, A.; Eisenhaure, T.; Schweitzer, L.D.; Hoover, P.; Parkinson, N.J.; Nachshon, A.; et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Forst, C.V.; Chou, T.-W.; Geber, A.; Wang, M.; Hamou, W.; Smith, M.; Sebra, R.; Zhang, B.; Zhou, B.; et al. Cell-to-Cell Variation in Defective Virus Expression and Effects on Host Responses during Influenza Virus Infection. mBio 2020, 11. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef]
- Hale, B.; Albrecht, R.A.; García-Sastre, A. Innate immune evasion strategies of influenza viruses. Future Microbiol. 2010, 5, 23–41. [Google Scholar] [CrossRef]
- Wilson, E.B.; Brooks, D.G. The Role of IL-10 in Regulating Immunity to Persistent Viral Infections. Curr. Top. Microbiol. Immunol. 2011, 350, 39–65. [Google Scholar]
- Ampomah, P.B.; Moraes, L.A.; Lukman, H.M.; Lim, L.H.K. Formyl peptide receptor 2 is regulated by RNA mimics and viruses through an IFN-β-STAT3-dependent pathway. FASEB J. 2018, 32, 1468–1478. [Google Scholar] [CrossRef]
- Arora, S.; Lim, W.; Bist, P.; Perumalsamy, R.; Lukman, H.M.; Li, F.; Welker, L.B.; Yan, B.; Sethi, G.; A Tambyah, P.; et al. Influenza A virus enhances its propagation through the modulation of Annexin-A1 dependent endosomal trafficking and apoptosis. Cell Death Differ. 2016, 23, 1243–1256. [Google Scholar] [CrossRef]
- Tran, A.T.; Cortens, J.P.; Du, Q.; Wilkins, J.A.; Coombs, K.M. Influenza Virus Induces Apoptosis via BAD-Mediated Mitochondrial Dysregulation. J. Virol. 2013, 87, 1049–1060. [Google Scholar] [CrossRef]
- Tripathi, S.; Batra, J.; Cao, W.; Sharma, K.; Patel, J.; Ranjan, P.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; et al. Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: Implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis. 2013, 4, e562. [Google Scholar] [CrossRef] [PubMed]
- Feizi, N.; Mehrbod, P.; Romani, B.; Soleimanjahi, H.; Bamdad, T.; Feizi, A.; Jazaeri, E.O.; Targhi, H.S.; Saleh, M.; Jamali, A.; et al. Autophagy induction regulates influenza virus replication in a time-dependent manner. J. Med. Microbiol. 2017, 66, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.K.; Ashour, J.; Karssemeijer, R.A.; Popp, M.W.; Avalos, A.M.; Barisa, M.; Altenburg, A.; Ingram, J.R.; Cragnolini, J.J.; Guo, C.; et al. Antigen-specific B-cell receptor sensitizes B cells to infection by influenza virus. Nature 2013, 503, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martínez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins. Viruses 2018, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.-L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef]
- Mendelson, M.; Tekoah, Y.; Zilka, A.; Gershoni-Yahalom, O.; Gazit, R.; Achdout, H.; Bovin, N.V.; Meningher, T.; Mandelboim, M.; Mandelboim, O.; et al. NKp46 O-Glycan Sequences That Are Involved in the Interaction with Hemagglutinin Type 1 of Influenza Virus. J. Virol. 2010, 84, 3789–3797. [Google Scholar] [CrossRef]
- Guo, H.; Kumar, P.; Malarkannan, S. Evasion of natural killer cells by influenza virus. J. Leukoc. Biol. 2010, 89, 189–194. [Google Scholar] [CrossRef]
- Li, T.; Wang, J.; Wang, Y.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Respiratory Influenza Virus Infection Induces Memory-like Liver NK Cells in Mice. J. Immunol. 2016, 198, 1242–1252. [Google Scholar] [CrossRef]
- Abdul-Careem, M.F.; Mian, M.F.; Yue, G.; Gillgrass, A.; Chenoweth, M.; Barra, N.G.; Chew, M.V.; Chan, T.; Al-Garawi, A.A.; Jordana, M.; et al. Critical Role of Natural Killer Cells in Lung Immunopathology During Influenza Infection in Mice. J. Infect. Dis. 2012, 206, 167–177. [Google Scholar] [CrossRef]
- Zhou, G.; Juang, S.W.W.; Kane, K.P. NK cells exacerbate the pathology of influenza virus infection in mice. Eur. J. Immunol. 2013, 43, 929–938. [Google Scholar] [CrossRef]
- Zhou, K.; Wang, J.; Li, A.; Zhao, W.; Wang, N.; Zhang, W.; Yan, J.; Gao, G.F.; Liu, W.; Fang, M. Swift and Strong NK Cell Responses Protect 129 Mice against High-Dose Influenza Virus Infection. J. Immunol. 2016, 196, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.; Jonsson, C.B. A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol. 2017, 8, 550. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Hyun, Y.-M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science 2015, 349, 4352. [Google Scholar] [CrossRef] [PubMed]
- GeurtsvanKessel, C.H.; Willart, M.A.; Van Rijt, L.S.; Muskens, F.; Kool, M.; Baas, C.; Thielemans, K.; Bennett, C.L.; Clausen, B.E.; Hoogsteden, H.C.; et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J. Exp. Med. 2008, 205, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Hintzen, G.; Ohl, L.; Del Rio, M.-L.; Rodriguez-Barbosa, J.-I.; Pabst, O.; Kocks, J.R.; Krege, J.; Hardtke, S.; Förster, R. Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J. Immunol. 2006, 177, 7346–7354. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-J.; Ishido, S.; Eisenlohr, L.C.; Roche, P.A. Activation of Dendritic Cells Alters the Mechanism of MHC Class II Antigen Presentation to CD4 T Cells. J. Immunol. 2020, 204, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Waithman, J.; Mintern, J.D. Dendritic cells and influenza A virus infection. Virulence 2012, 3, 603–608. [Google Scholar] [CrossRef]
- Takeshita, F.; Tanaka, T.; Matsuda, T.; Tozuka, M.; Kobiyama, K.; Saha, S.; Matsui, K.; Ishii, K.J.; Coban, C.; Akira, S.; et al. Toll-Like Receptor Adaptor Molecules Enhance DNA-Raised Adaptive Immune Responses against Influenza and Tumors through Activation of Innate Immunity. J. Virol. 2006, 80, 6218–6224. [Google Scholar] [CrossRef]
- Schulz, O.; Diebold, S.S.; Chen, M.S.; Näslund, T.I.; Nolte, M.; Alexopoulou, L.; Azuma, Y.-T.; Flavell, R.A.; Liljestrom, P.; E Sousa, C.R. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 2007, 178, 3368–3372. [Google Scholar] [CrossRef]
- Poux, C.; Dondalska, A.; Bergenstråhle, J.; Pålsson, S.; Contreras, V.; Arasa, C.; Järver, P.; Albert, J.; Busse, D.; Legrand, R.; et al. A Single-Stranded Oligonucleotide Inhibits Toll-Like Receptor 3 Activation and Reduces Influenza A (H1N1) Infection. Front. Immunol. 2019, 10, 2161. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Cader, M.S.; Senapathi, U.D.S.; Nagy, E.; Sharif, S.; Abdul-Careem, M.F. Antiviral response elicited against avian influenza virus infection following activation of toll-like receptor (TLR)7 signaling pathway is attributable to interleukin (IL)-1β production. BMC Res. Notes 2018, 11, 859. [Google Scholar] [CrossRef] [PubMed]
- Stegemann-Koniszewski, S.; Behrens, S.; Boehme, J.D.; Hochnadel, I.; Riese, P.; Guzmán, C.A.; Kröger, A.; Schreiber, J.; Gunzer, M.; Bruder, D. Respiratory Influenza A Virus Infection Triggers Local and Systemic Natural Killer Cell Activation via Toll-Like Receptor 7. Front. Immunol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Jeisy-Scott, V.; Kim, J.H.; Davis, W.G.; Cao, W.; Katz, J.M.; Sambhara, S. TLR7 Recognition Is Dispensable for Influenza Virus A Infection but Important for the Induction of Hemagglutinin-Specific Antibodies in Response to the 2009 Pandemic Split Vaccine in Mice. J. Virol. 2012, 86, 10988–10998. [Google Scholar] [CrossRef]
- Walsh, K.B.; Teijaro, J.R.; Zuniga, E.I.; Welch, M.J.; Fremgen, D.M.; Blackburn, S.D.; Von Tiehl, K.F.; Wherry, E.J.; Flavell, R.A.; Oldstone, M.B.A. Toll-like Receptor 7 Is Required for Effective Adaptive Immune Responses that Prevent Persistent Virus Infection. Cell Host Microbe 2012, 11, 643–653. [Google Scholar] [CrossRef]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef]
- Jahan, A.S.; Biquand, E.; Muñoz-Moreno, R.; Le Quang, A.; Mok, C.K.-P.; Wong, H.H.; Teo, Q.W.; Valkenburg, S.A.; Chin, A.W.; Poon, L.L.M.; et al. OTUB1 Is a Key Regulator of RIG-I-Dependent Immune Signaling and Is Targeted for Proteasomal Degradation by Influenza A NS1. Cell Rep. 2020, 30, 1570–1584. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.-S.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.; Tate, M.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.; Mansell, A. Activation of the NLRP3 Inflammasome by IAV Virulence Protein PB1-F2 Contributes to Severe Pathophysiology and Disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed]
- Sarvestani, S.T.; McAuley, J.L. The role of the NLRP3 inflammasome in regulation of antiviral responses to influenza A virus infection. Antivir. Res. 2017, 148, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Liu, B.; Bao, L.; Lv, Q.; Li, F.; Li, H.; An, Y.; Zhang, X.; Cao, B.; Wang, C. Delayed oseltamivir plus sirolimus treatment attenuates H1N1 virus-induced severe lung injury correlated with repressed NLRP3 inflammasome activation and inflammatory cell infiltration. PLoS Pathog. 2018, 14, e1007428. [Google Scholar] [CrossRef]
- Randall, R.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Mordstein, M.; Kochs, G.; Dumoutier, L.; Renauld, J.C.; Paludan, S.R.; Klucher, K.; Staeheil, P. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog 2008, 4, e1000151. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Crotta, S.; Davidson, S.; Mahlakõiv, T.; Desmet, C.J.; Buckwalter, M.R.; Albert, M.L.; Staeheli, P.; Wack, A. Type I and Type III Interferons Drive Redundant Amplification Loops to Induce a Transcriptional Signature in Influenza-Infected Airway Epithelia. PLoS Pathog. 2013, 9, e1003773. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.-E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890. [Google Scholar] [CrossRef]
- Hemann, E.A.; Green, R.; Turnbull, J.B.; Langlois, R.A.; Savan, R.; Gale, M. Interferon-λ modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nat. Immunol. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Xiao, H.; Killip, M.J.; Staeheli, P.; Randall, R.; Jackson, D. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J. Virol. 2013, 87, 13053–13058. [Google Scholar] [CrossRef] [PubMed]
- Mänz, B.; Dornfeld, D.; Götz, V.; Zell, R.; Zimmermann, P.; Haller, O.; Kochs, G.; Schwemmle, M. Pandemic Influenza A Viruses Escape from Restriction by Human MxA through Adaptive Mutations in the Nucleoprotein. PLoS Pathog. 2013, 9, e1003279. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef]
- Nordmann, A.; Wixler, L.; Boergeling, Y.; Wixler, V.; Ludwig, S. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 2012, 26, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, J.; Wang, S.; Wu, F.; Chen, Z.; Li, C.; Cheng, G.; Qin, F.X.-F. Inhibition of Influenza A Virus Replication by TRIM14 via Its Multifaceted Protein-Protein Interaction with NP. Front. Microbiol. 2019, 10, 344. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 Inhibits Influenza A Virus Infection by Targeting the Viral Nucleoprotein for Degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef]
- Patil, G.; Zhao, M.; Song, K.; Hao, W.; Bouchereau, D.; Wang, L.; Li, S. TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Influenza A Virus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Shim, J.M.; Kim, J.; Tenson, T.; Min, J.-Y.; Kainov, D. Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis. Viruses 2017, 9, 223. [Google Scholar] [CrossRef]
- Killip, M.J.; Fodor, E.; Randall, R. Influenza virus activation of the interferon system. Virus Res. 2015, 209, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Vijayan, M.; Pritzl, C.J.; Fuchs, S.Y.; McDermott, A.B.; Hahm, B. Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1. J. Virol. 2015, 90, 2403–2417. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Wolf, J.J.; Sun, C.; Xu, M.; Studstill, C.J.; Chen, J.; Ngo, H.; Zhu, H.; Hahm, B. PARP1 Enhances Influenza A Virus Propagation by Facilitating Degradation of Host Type I Interferon Receptor. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza Virus Protein PB1-F2 Inhibits the Induction of Type I Interferon by Binding to MAVS and Decreasing Mitochondrial Membrane Potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef]
- Park, E.; Byun, Y.H.; Park, S.; Jang, Y.H.; Han, W.; Won, J.; Cho, K.C.; Kim, D.H.; Lee, A.R.; Shin, G.-C.; et al. Co-degradation of interferon signaling factor DDX3 by PB1-F2 as a basis for high virulence of 1918 pandemic influenza. EMBO J. 2019, 38, e99475. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2020, 11. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, H.; Jiang, C.C.; Fang, L.; Chen, C.; Zhang, X.D.; Jiang, Z.W. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int. J. Mol. Med. 2014, 34, 772–781. [Google Scholar] [CrossRef]
- Zhong, L.; Shu, W.; Dai, W.; Gao, B.; Xiong, S. Reactive Oxygen Species-Mediated c-Jun NH2-Terminal Kinase Activation Contributes to Hepatitis B Virus X Protein-Induced Autophagy via Regulation of the Beclin-1/Bcl-2 Interaction. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Li, D.-D.; Wang, L.-L.; Deng, R.; Tang, J.; Shen, Y.; Guo, J.-F.; Wang, Y.; Xia, L.-P.; Feng, G.-K.; Liu, Q.Q.; et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009, 28, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chi, X.; Wang, S.; Qi, B.; Yu, X.; Chen, J.-L. The Regulation of Autophagy by Influenza A Virus. BioMed Res. Int. 2014, 2014, 498083. [Google Scholar] [CrossRef] [PubMed]
- Sparrer, K.M.J.; Gableske, S.; Zurenski, M.A.; Parker, Z.M.; Full, F.; Baumgart, G.J.; Kato, J.; Pacheco-Rodriguez, G.; Liang, C.; Pornillos, O.; et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat. Microbiol. 2017, 2, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, T.; Osari, S.; Nagata, K.; Kawaguchi, A. Influenza A Virus NS1 Protein Suppresses JNK1-Dependent Autophagosome Formation Mediated by Rab11a Recycling Endosomes. Front. Microbiol. 2018, 9, 3120. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Sun, Y.; Sun, J.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. Role of TGF-β-activated kinase 1 (TAK1) activation in H5N1 influenza A virus-induced c-Jun terminal kinase activation and virus replication. Virology 2019, 537, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hong, M.J.; Sun, H.; Wang, L.; Shi, X.; Gilbert, B.E.; Corry, D.B.; Kheradmand, F.; Wang, J. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat. Med. 2014, 20, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Gannagé, M.; Dormann, R.; Albrecht, R.A.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza A Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LIR motif in influenza A virus M2 is required for virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef]
- Ren, Y.; Li, C.; Feng, L.; Pan, W.; Li, L.; Wang, Q.; Li, J.; Li, N.; Han, L.; Zheng, X.; et al. Proton Channel Activity of Influenza A Virus Matrix Protein 2 Contributes to Autophagy Arrest. J. Virol. 2015, 90, 591–598. [Google Scholar] [CrossRef]
- Yu, T.; Ding, Y.; Zhang, Y.; Liu, Y.; Li, Y.; Lei, J.; Zhou, J.; Song, S.; Hu, B. Circular RNA GATAD2A promotes H1N1 replication through inhibiting autophagy. Vet. Microbiol. 2019, 231, 238–245. [Google Scholar] [CrossRef]
- Ampomah, P.B.; Lim, L.H.K. Influenza A virus-induced apoptosis and virus propagation. Apoptosis 2020, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.; Castrucci, M.R.; Padrick, R.C.; Bradley, L.M.; Topham, D.J. Antigen-specific and non-specific CD4+ T cell recruitment and proliferation during influenza infection. Virology 2005, 340, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L.; McKinstry, K.; Strutt, T.M. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Lindell, D.M.; Berlin, A.A.; Morris, S.B.; Shanley, T.; Hershenson, M.B.; Lukacs, N.W. IL-17–Induced Pulmonary Pathogenesis during Respiratory Viral Infection and Exacerbation of Allergic Disease. Am. J. Pathol. 2011, 179, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Almansa, R.; Socias, L.; Ramirez, P.; Martin-Loeches, I.; Vallés, J.; Loza, A.; Jordi, R.; David, J.K.; Cristobal, L.; Jesús, B.; et al. Imbalanced pro- and anti-Th17 responses (IL-17/granulocyte colony-stimulating factor) predict fatal outcome in 2009 pandemic influenza. Crit. Care 2011, 15, 448. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Molony, R.D.; Kudo, E.; Sidorov, S.; Kong, Y.; Dixit, V.D.; Iwasaki, A. Ketogenic diet activates protective γδ T cell responses against influenza virus infection. Sci. Immunol. 2019, 4, 2026. [Google Scholar] [CrossRef]
- Hornick, E.E.; Zacharias, Z.R.; Legge, K.L. Kinetics and Phenotype of the CD4 T Cell Response to Influenza Virus Infections. Front. Immunol. 2019, 10, 2351. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Turner, S.J. T cell mediated immunity to influenza: Mechanisms of viral control. Trends Immunol. 2014, 35, 396–402. [Google Scholar] [CrossRef]
- Andrade, F. Non-cytotoxic antiviral activities of granzymes in the context of the immune antiviral state. Immunol. Rev. 2010, 235, 128–146. [Google Scholar] [CrossRef]
- Allie, S.R.; Randall, T.D. Pulmonary immunity to viruses. Clin. Sci. 2017, 131, 1737–1762. [Google Scholar] [CrossRef]
- Jia, R.; Liu, S.; Xu, J.; Liang, X. IL16 deficiency enhances Th1 and cytotoxic T lymphocyte response against influenza A virus infection. Biosci. Trends 2020, 13, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Grant, E.J.; Quiñones-Parra, S.M.; Clemens, E.B.; Kedzierska, K. Human influenza viruses and CD8 + T cell responses. Curr. Opin. Virol. 2016, 16, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Pizzolla, A.; Nguyen, T.H.O.; Smith, J.M.; Brooks, A.G.; Kedzierska, K.; Heath, W.R.; Reading, P.; Wakim, L.M. Resident memory CD8+T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. 2017, 2, 6970. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.M.; Klenerman, P.; et al. Autophagy is a critical regulator of memory CD8+ T cell formation. ELife 2014, 3, e03706. [Google Scholar] [CrossRef]
- Cullen, J.G.; McQuilten, H.A.; Quinn, K.M.; Olshansky, M.; Russ, B.E.; Morey, A.; Wei, S.; Prier, J.E.; La Gruta, N.L.; Doherty, P.C.; et al. CD4+T help promotes influenza virus-specific CD8+T cell memory by limiting metabolic dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 4481–4488. [Google Scholar] [CrossRef]
- Rangel-Moreno, J.; Carragher, D.; Misra, R.S.; Kusser, K.; Hartson, L.; Moquin, A.; Lund, F.E.; Randall, T.D. B cells promote resistance to heterosubtypic strains of influenza via multiple mechanisms1. J. Immunol. 2008, 180, 454–463. [Google Scholar] [CrossRef]
- Seibert, C.W.; Rahmat, S.; Krause, J.C.; Eggink, D.; Albrecht, R.A.; Goff, P.H.; Krammer, F.; Duty, J.A.; Bouvier, N.; García-Sastre, A.; et al. Recombinant IgA Is Sufficient To Prevent Influenza Virus Transmission in Guinea Pigs. J. Virol. 2013, 87, 7793–7804. [Google Scholar] [CrossRef]
- Hervé, P.-L.; Lorin, V.; Jouvion, G.; Da Costa, B.; Escriou, N. Addition of N-glycosylation sites on the globular head of the H5 hemagglutinin induces the escape of highly pathogenic avian influenza A H5N1 viruses from vaccine-induced immunity. Virology 2015, 486, 134–145. [Google Scholar] [CrossRef]
- A Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Kapranos, N.; Kontogeorgos, G. Bcl-2 Gene Family in Endocrine Pathology: A Review. Endocr. Pathol. 2000, 11, 205–214. [Google Scholar]
- Brüne, B. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ. 2003, 10, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.D.; Schmitz, I.; Chen, M.; Wang, J. Promotion of Caspase Activation by Caspase-9-mediated Feedback Amplification of Mitochondrial Damage. J. Clin. Cell. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Strasser, A.; Jost, P.J.; Nagata, S. The Many Roles of FAS Receptor Signaling in the Immune System. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef]
- Wajant, H. The Fas Signaling Pathway: More Than a Paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral Control of Mitochondrial Apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef]
- Zhirnov, O.P.; Klenk, H.-D. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis 2007, 12, 1419–1432. [Google Scholar] [CrossRef]
- Nailwal, H.; Sharma, S.; Mayank, A.K.; Lal, S.K. The nucleoprotein of influenza A virus induces p53 signaling and apoptosis via attenuation of host ubiquitin ligase RNF43. Cell Death Dis. 2015, 6, e1768. [Google Scholar] [CrossRef]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef]
- Halder, U.C.; Bagchi, P.; Chattopadhyay, S.; Dutta, D.; Chawla-Sarkar, M. Cell death regulation during influenza A virus infection by matrix (M1) protein: A model of viral control over the cellular survival pathway. Cell Death Dis. 2011, 2, e197. [Google Scholar] [CrossRef]
- Schultz-Cherry, S.; Dybdahl-Sissoko, N.; Neumann, G.; Kawaoka, Y.; Hinshaw, V.S. Influenza Virus NS1 Protein Induces Apoptosis in Cultured Cells. J. Virol. 2001, 75, 7875–7881. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Killip, M.J.; Galloway, C.S.; Russell, R.J.; Randall, R.E. Loss of function of the influenza A virus NS1 protein promotes apoptosis but this is not due to a failure to activate phosphatidylinositol 3-kinase (PI3K). Virology 2010, 396, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun. 2014, 5, 3864. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Rune, H.; Andreas, W. IFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment. EMBO Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hirohama, M.; Noguchi, M.; Nagata, K.; Kawaguchi, A. Influenza A Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Grodeland, G.; Fossum, E.; Bogen, B. Targeting of HA to chemokine receptors induces strong and cross-reactive T cell responses after DNA vaccination in pigs. Vaccine 2020, 38, 1280–1285. [Google Scholar] [CrossRef]
- Zacharias, Z.R.; Ross, K.A.; Hornick, E.E.; Goodman, J.T.; Narasimhan, B.; Waldschmidt, T.; Legge, K.L. Polyanhydride Nanovaccine Induces Robust Pulmonary B and T Cell Immunity and Confers Protection Against Homologous and Heterologous Influenza A Virus Infections. Front. Immunol. 2018, 9, 1953. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J. Influenza neuraminidase inhibitors: Antiviral action and mechanisms of resistance. Influenza Other Respir. Viruses 2013, 7, 25–36. [Google Scholar] [CrossRef]
- O’Hanlon, R.; Shaw, M. Baloxavir marboxil: The new influenza drug on the market. Curr. Opin. Virol. 2019, 35, 14–18. [Google Scholar] [CrossRef]
- Vanderlinden, E.; Naesens, L. Emerging Antiviral Strategies to Interfere with Influenza Virus Entry. Med. Res. Rev. 2014, 34, 301–339. [Google Scholar] [CrossRef]
- Song, G.; Yang, S.; Zhang, W.; Cao, Y.; Wang, P.; Ding, N.; Zhang, Z.; Guo, Y.; Li, Y. Discovery of the First Series of Small Molecule H5N1 Entry Inhibitors. J. Med. Chem. 2009, 52, 7368–7371. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, N.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B 2017, 93, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, F.; Meng, L.; Sun, J.; Su, Y.; Shao, L.; Zhou, D.; Yu, F. Synthesis, Structure Activity Relationship and Anti-influenza A Virus Evaluation of Oleanolic Acid-Linear Amino Derivatives. Chem. Pharm. Bull. 2019, 67, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Meng, L.; Sun, J.; Li, W.; Shao, L.; Chen, K.; Zhou, D.; Yang, F.; Yu, F. Design, synthesis of oleanolic acid-saccharide conjugates using click chemistry methodology and study of their anti-influenza activity. Eur. J. Med. Chem. 2019, 182, 111622. [Google Scholar] [CrossRef]
- Meng, L.; Su, Y.; Yang, F.; Xiao, S.; Yin, Z.; Liu, J.; Zhong, J.; Zhou, D.; Yu, F. Design, synthesis and biological evaluation of amino acids-oleanolic acid conjugates as influenza virus inhibitors. Bioorg. Med. Chem. 2019, 27, 115147. [Google Scholar] [CrossRef]
- Omi, J.; Watanabe-Takahashi, M.; Igai, K.; Shimizu, E.; Tseng, C.-Y.; Miyasaka, T.; Waku, T.; Hama, S.; Nakanishi, R.; Goto, Y.; et al. The inducible amphisome isolates viral hemagglutinin and defends against influenza A virus infection. Nat. Commun. 2020, 11, 162. [Google Scholar] [CrossRef]
- Clark, M.P.; Ledeboer, M.W.; Davies, I.; Byrn, R.A.; Jones, S.M.; Perola, E.; Tsai, A.; Jacobs, M.; Nti-Addae, K.; Bandarage, U.K.; et al. Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2. J. Med. Chem. 2014, 57, 6668–6678. [Google Scholar] [CrossRef]
- Jones, J.C.; Marathe, B.M.; Lerner, C.; Kreis, L.; Gasser, R.; Pascua, P.N.Q.; Najera, I.; Govorkova, E.A. A Novel Endonuclease Inhibitor Exhibits Broad-Spectrum Anti-Influenza Virus ActivityIn Vitro. Antimicrob. Agents Chemother. 2016, 60, 5504–5514. [Google Scholar] [CrossRef]
- Song, M.-S.; Kumar, G.; Shadrick, W.R.; Zhou, W.; Jeevan, T.; Li, Z.; Slavish, P.J.; Fabrizio, T.; Yoon, S.-W.; Webb, T.R.; et al. Identification and characterization of influenza variants resistant to a viral endonuclease inhibitor. Proc. Natl. Acad. Sci. USA 2016, 113, 3669–3674. [Google Scholar] [CrossRef]
- Massari, S.; Goracci, L.; DeSantis, J.; Tabarrini, O. Polymerase Acidic Protein–Basic Protein 1 (PA–PB1) Protein-Protein Interaction as a Target for Next-Generation Anti-influenza Therapeutics. J. Med. Chem. 2016, 59, 7699–7718. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Fan, W.; Zhang, S.; Jiao, P.; Shang, Y.; Cui, L.; Mahesutihan, M.; Li, J.; Wang, D.; Gao, G.F.; et al. Naproxen Exhibits Broad Anti-influenza Virus Activity in Mice by Impeding Viral Nucleoprotein Nuclear Export. Cell Rep. 2019, 27, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Pickens, J.A.; Tripp, R.A. Verdinexor Targeting of CRM1 is a Promising Therapeutic Approach against RSV and Influenza Viruses. Viruses 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S. Treating Influenza Infection, From Now and Into the Future. Front. Immunol. 2018, 9, 1946. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Xu, Z.; Cao, Y. Host–Virus Interaction: How Host Cells Defend against Influenza A Virus Infection. Viruses 2020, 12, 376. https://doi.org/10.3390/v12040376
Zhang Y, Xu Z, Cao Y. Host–Virus Interaction: How Host Cells Defend against Influenza A Virus Infection. Viruses. 2020; 12(4):376. https://doi.org/10.3390/v12040376
Chicago/Turabian StyleZhang, Yun, Zhichao Xu, and Yongchang Cao. 2020. "Host–Virus Interaction: How Host Cells Defend against Influenza A Virus Infection" Viruses 12, no. 4: 376. https://doi.org/10.3390/v12040376
APA StyleZhang, Y., Xu, Z., & Cao, Y. (2020). Host–Virus Interaction: How Host Cells Defend against Influenza A Virus Infection. Viruses, 12(4), 376. https://doi.org/10.3390/v12040376