Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins

 , , ,
, , ,
Abstract
1. Importance
2. Introduction
3. Materials and Methods
3.1. Protein Sequence Data Collection and Analysis
3.2. Structural Characterization of Protein and Protein Complexes
3.3. Mapping of Functional Regions and Evolutionary Conservation
3.4. Inferring Intra-Viral and Virus–Host Protein–Protein Interaction Networks
4. Results
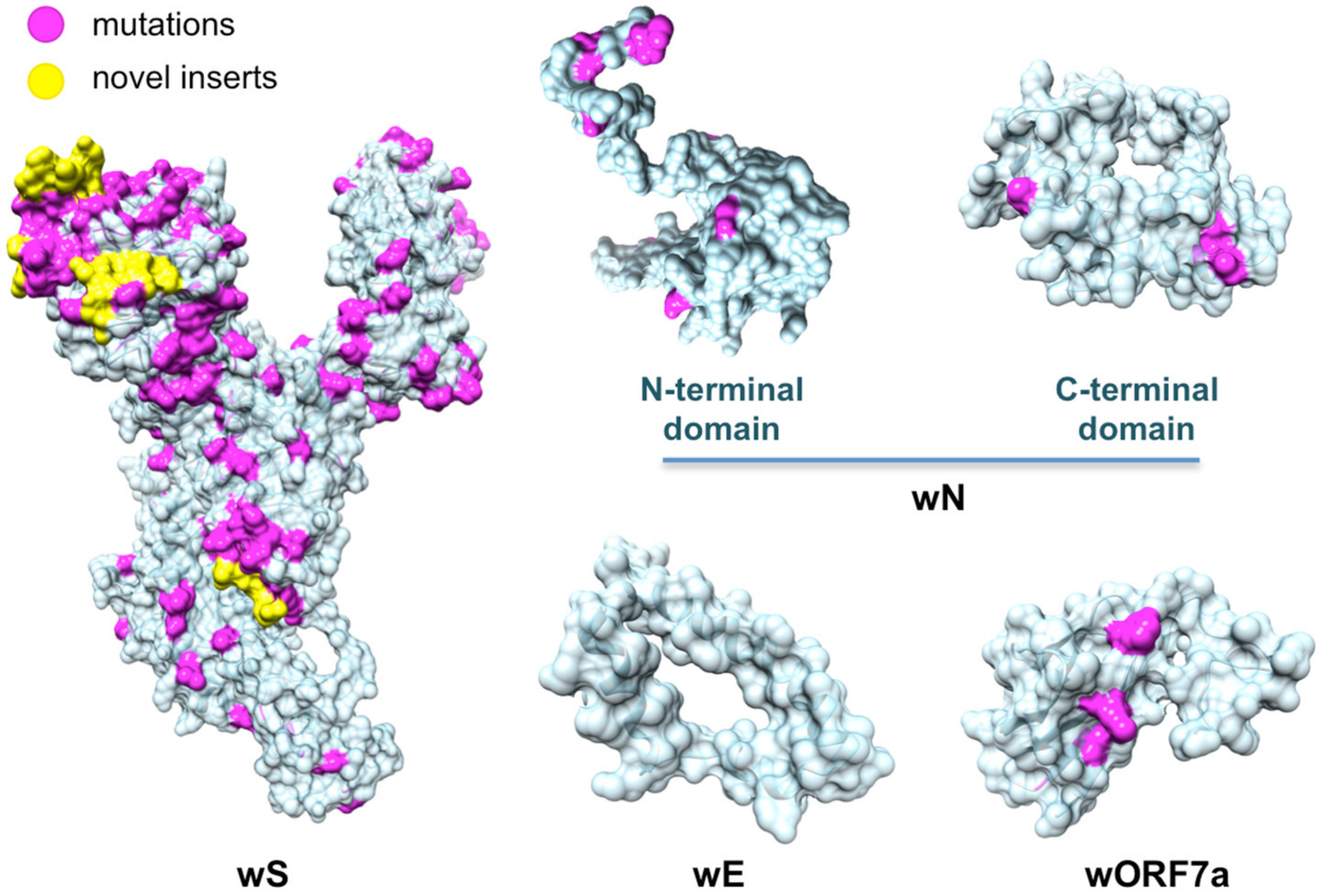
4.1. Comparative Analysis of SARS-CoV-2 Proteins With the Evolutionary Related Coronavirus Proteins Reveals Unevenly Distributed Large Genomic Insertions
4.2. Three Recent Strains of Bat SARS-like Coronavirus from 2013, 2015 and 2017 Share Extremely High Proteome Similarity with SARS-CoV-2
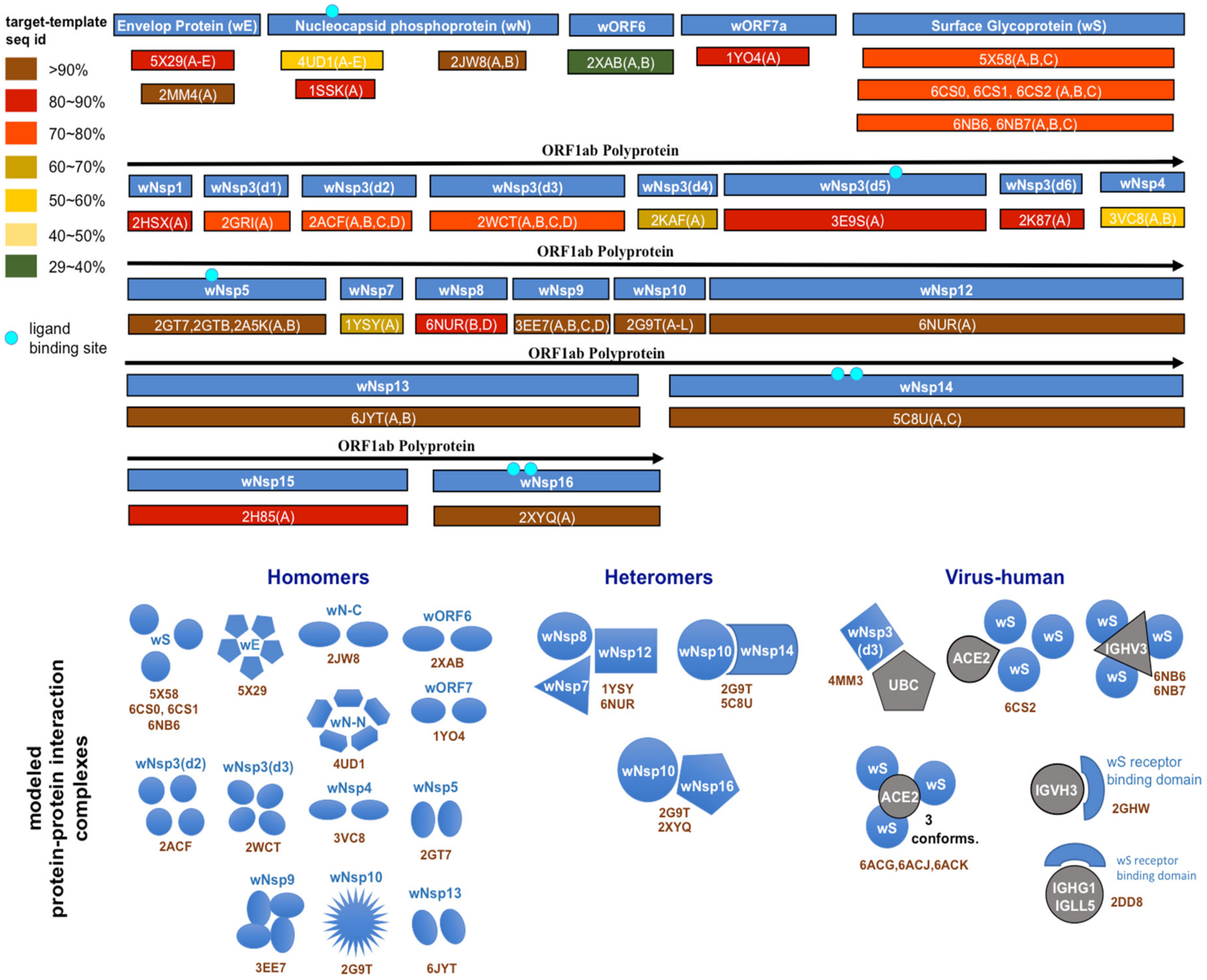
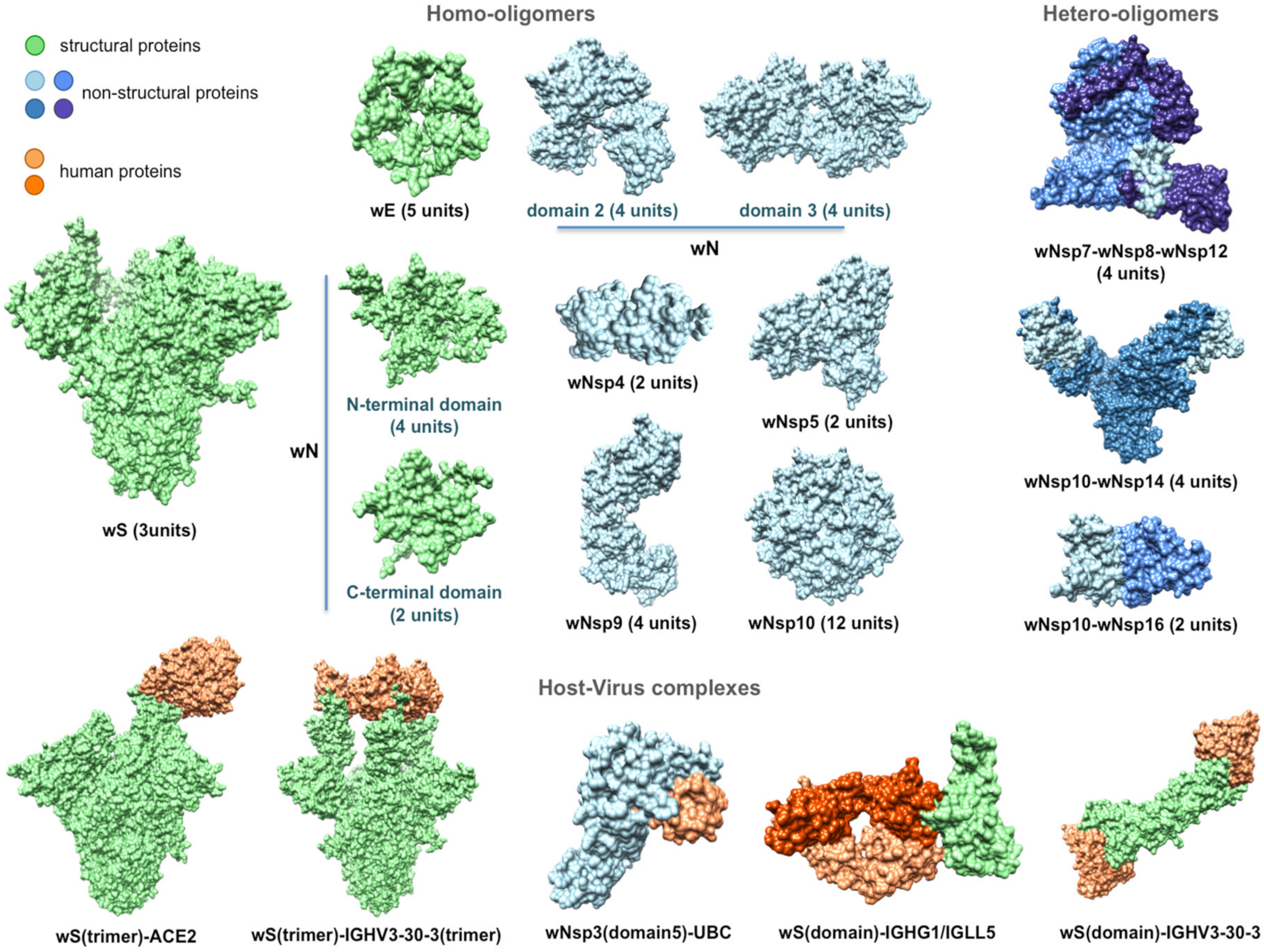
4.3. Structural Genomics and Interactomics Analysis of SARS-CoV-2
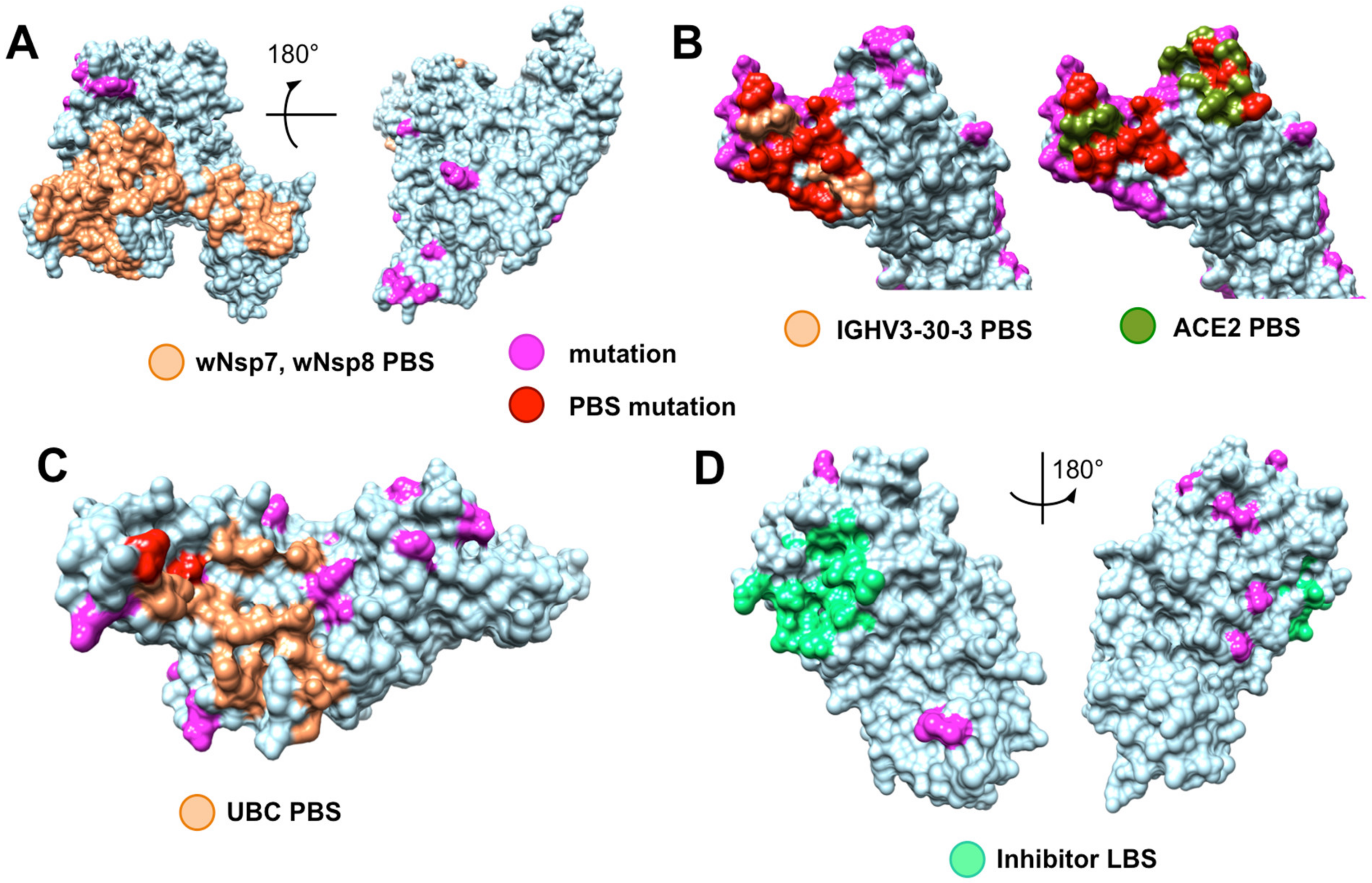
4.4. Evolutionary Conservation and Divergence of Functional Regions of SARS-CoV-2
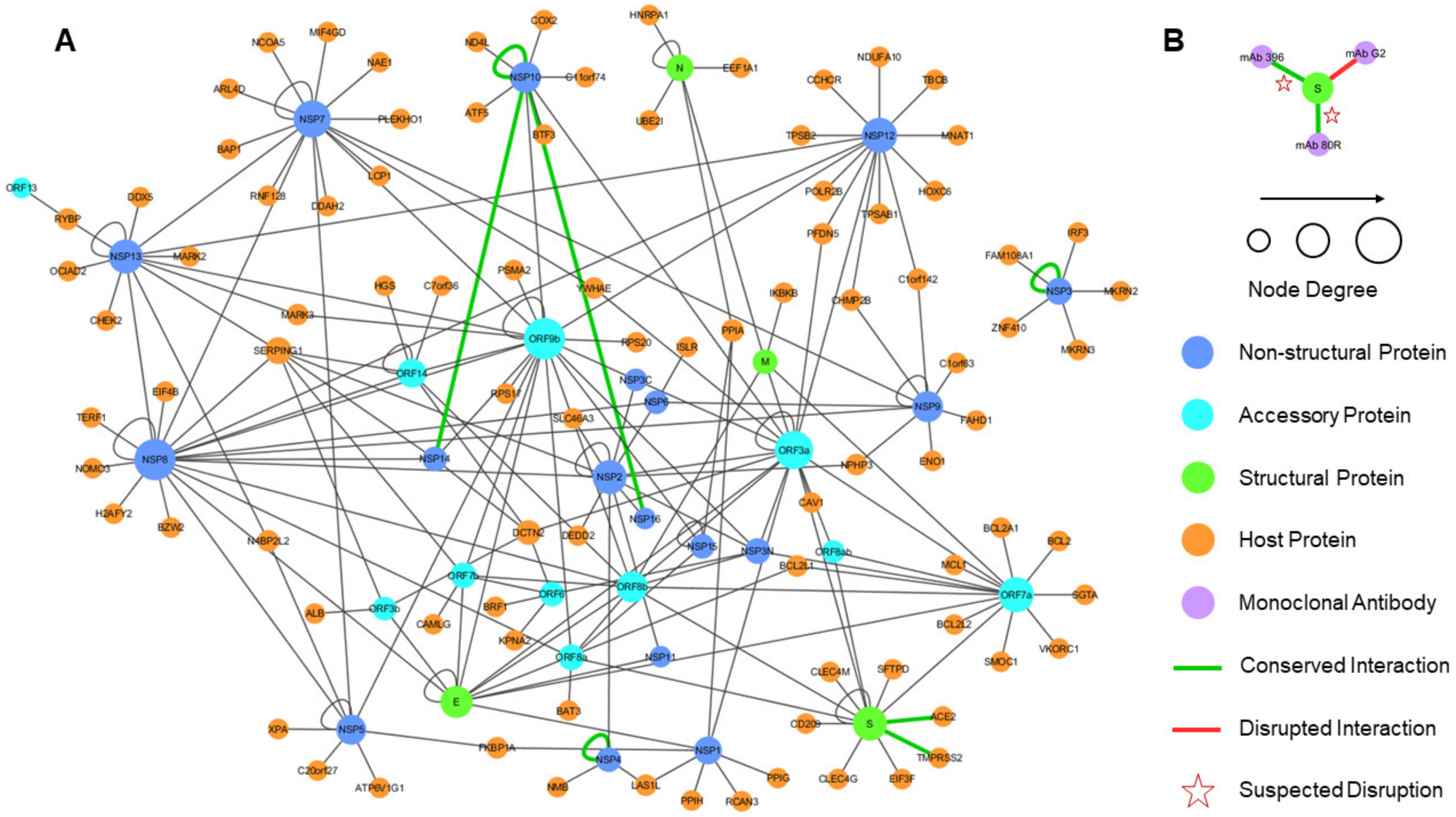
4.5. Joint Intra-Viral and Human–Virus Protein–Protein Interaction Network for SARS-CoV Indicates Potential System-Wide Roles of SARS-CoV-2 Proteins
5. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing SARS-CoV-2 epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.J. Designing tomorrow’s vaccines. N. Engl. J. Med. 2013, 368, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Plotkin, S.L. The development of vaccines: How the past led to the future. Nat. Rev. Microbiol. 2011, 9, 889–893. [Google Scholar] [CrossRef]
- Martin, J.E.; Louder, M.K.; Holman, L.A.; Gordon, I.J.; Enama, M.E.; Larkin, B.D.; Andrews, C.A.; Vogel, L.; Koup, R.A.; Roederer, M.; et al. A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a Phase I clinical trial. Vaccine 2008, 26, 6338–6343. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.-W.; Azhar, E.I.; Hui, D.S.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- Zumla, A.; Memish, Z.A.; Hui, D.S.; Perlman, S. Vaccine against Middle East respiratory syndrome coronavirus. Lancet Infect. Dis. 2019, 19, 1054–1055. [Google Scholar] [CrossRef]
- Cotten, M.; Watson, S.; Kellam, P.; Al-Rabeeah, A.A.; Makhdoom, H.Q.; Assiri, A.; Al-Tawfiq, J.A.; AlHakeem, R.F.; Madani, H.; AlRabiah, F.A.; et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive genomic study. Lancet 2013, 382, 1993–2002. [Google Scholar] [CrossRef]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Genet. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Dyall, J.; Coleman, C.; Hart, B.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of Clinically Developed Drugs for Treatment of Middle East Respiratory Syndrome Coronavirus Infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Ann. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Luk, H.K.; Li, X.; Fung, J.; Lau, S.K.P.; Woo, P.C.Y. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019, 71, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Physiological and molecular triggers for SARS-CoV membrane fusion and entry into host cells. Virology 2018, 517, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLOS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.; Tortorici, M.A.; Bosch, B.-J.; Frenz, B.; Rottier, P.J.M.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Cho, C.C.; Lin, M.H.; Chuang, C.Y.; Hsu, C.H. Macro Domain from Middle East. Respiratory Syndrome Coronavirus (MERS-CoV) Is an Efficient ADP-ribose Binding Module CRYSTAL STRUCTURE AND BIOCHEMICAL STUDIES. J. Biol. Chem. 2016, 291, 4894–4902. [Google Scholar] [CrossRef]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2016, 27, 119–129. [Google Scholar] [CrossRef]
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M.; Tomar, S.; et al. Discovery, Synthesis, And Structure-Based Optimization of a Series ofN-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) Acetamides (ML188) as Potent Noncovalent Small Molecule Inhibitors of the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) 3CL Protease. J. Med. Chem. 2013, 56, 534–546. [Google Scholar] [PubMed]
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef] [PubMed]
- Kankanamalage, A.C.G.; Kim, Y.; Damalanka, V.C.; Rathnayake, A.D.; Fehr, A.R.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Lushington, G.; Perlman, S.; et al. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 2018, 150, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Wang, N.; Pallesen, J.; Wrapp, D.; Turner, H.L.; Cottrell, C.A.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342–2349. [Google Scholar] [CrossRef]
- Li, Y.; Wan, Y.; Liu, P.; Zhao, J.; Lu, G.; Qi, J.; Wang, Q.; Lu, X.; Wu, Y.; Liu, W.; et al. A humanized neutralizing antibody against MERS-CoV targeting the receptor-binding domain of the spike protein. Cell Res. 2015, 25, 1237–1249. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Báez-Santos, Y.M.; Baker, S.C.; Mesecar, A.D. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating Activity of SARS-CoV Papain-Like Protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, Y.; Hattori, Y.; Kobayashi, K.; Teruya, K.; Sanjoh, A.; Nakagawa, A.; Yamashita, E.; Akaji, K. Fused-ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors. Bioorganic Med. Chem. 2015, 23, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Lou, Z.; Sun, F.; Zhai, Y.; Yang, H.; Zhang, R.; Joachimiak, A.; Zhang, X.C.; Bartlam, M.; Rao, Z. Dodecamer structure of severe acute respiratory syndrome coronavirus non-structural protein nsp10. J. Virol. 2006, 80, 7902–7908. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Rosen, O.; Wang, L.; Turner, H.L.; Stevens, L.J.; Corbett, K.S.; Bowman, C.A.; Pallesen, J.; Shi, W.; Zhang, Y.; et al. Structural Definition of a Neutralization-Sensitive Epitope on the MERS-CoV S1-NTD. Cell Rep. 2019, 28, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Sali, A.; Sánchez, R.; Melo, F.; Sali, A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Almo, S.C.; Bonanno, J.B.; Capel, M.; Chance, M.R.; Gaasterland, T.; Lin, D.; Sali, A.; Studier, F.W.; Swaminathan, S. Structural genomics: Beyond the Human Genome Project. Nat. Genet. 1999, 23, 151–157. [Google Scholar] [CrossRef]
- Yan, L.; Velikanov, M.; Flook, P.; Zheng, W.; Szalma, S.; Kahn, S. Assessment of putative protein targets derived from the SARS genome. FEBS Lett. 2003, 554, 257–263. [Google Scholar] [CrossRef]
- Wichapong, K.; Pianwanit, S.; Sippl, W.; Kokpol, S. Homology modeling and molecular dynamics simulations of Dengue virus NS2B/NS3 protease: Insight into molecular interaction. J. Mol. Recognit. 2009, 23, 283–300. [Google Scholar] [CrossRef]
- Ekins, S.; Liebler, J.; Neves, B.J.; Lewis, W.G.; Coffee, M.; Bienstock, R.; Southan, C.; Andrade, C.H. Illustrating and homology modeling the proteins of the Zika virus. F1000 Res. 2016, 5, 275. [Google Scholar] [CrossRef]
- Prabakaran, P.; Xiao, X.; Dimitrov, D.S. A model of the ACE2 structure and function as a SARS-CoV receptor. Biochem. Biophys. Res. Commun. 2004, 314, 235–241. [Google Scholar] [CrossRef]
- Davis, F.P.; Barkan, D.T.; Eswar, N.; McKerrow, J.H.; Sali, A. Host–pathogen protein interactions predicted by comparative modeling. Protein Sci. 2007, 16, 2585–2596. [Google Scholar] [CrossRef]
- Russell, R.B.; Alber, F.; Aloy, P.; Davis, F.P.; Korkin, D.; Pichaud, M.; Topf, M.; Sali, A. A structural perspective on protein–protein interactions. Curr. Opin. Struct. Boil. 2004, 14, 313–324. [Google Scholar] [CrossRef]
- Zhang, Q.C.; Petrey, N.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, M.; Hunter, T.; et al. Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Orry, A.J.W.; Murgolo, N.J.; Czarniecki, M.F.; Kocsi, S.A.; Hawes, B.E.; O’Neill, K.A.; Hine, H.; Burton, M.S.; Voigt, J.H.; et al. Discovery of Novel Chemotypes to a G-Protein-Coupled Receptor through Ligand-Steered Homology Modeling and Structure-Based Virtual Screening. J. Med. Chem. 2008, 51, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Q.; Du, Q.-S.; Chou, K.-C. Study of drug resistance of chicken influenza A virus (H5N1) from homology-modeled 3D structures of neuraminidases. Biochem. Biophys. Res. Commun. 2007, 354, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, Y.; Raimondo, M.; Redfern, O.; Watson, J.; Frishman, D.; Linial, M.; Orengo, C.; Thornton, J.M.; Tramontano, A. Protein function annotation by homology-based inference. Genome Boil. 2009, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Durmuş, S.; Ulgen, K.O. Comparative interactomics for virus-human protein-protein interactions: DNA viruses versus RNA viruses. FEBS Open Bio 2017, 7, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; He, L.; Wang, Y. Prediction of GCRV virus-host protein interactome based on structural motif-domain interactions. BMC Bioinform. 2017, 18, 145. [Google Scholar] [CrossRef]
- Vidalain, P.-O.; Tangy, F. Virus-host protein interactions in RNA viruses. Microbes Infect. 2010, 12, 1134–1143. [Google Scholar] [CrossRef]
- Lasso, G.; Mayer, S.V.; Winkelmann, E.R.; Chu, T.; Elliot, O.; Patino-Galindo, J.A.; Park, K.; Rabadan, R.; Honig, B.; Shapira, S.D. A Structure-Informed Atlas of Human-Virus Interactions. Cell 2019, 178, 1526. [Google Scholar] [CrossRef]
- Warren, S.; Wan, X.-F.; Conant, G.C.; Korkin, D. Extreme Evolutionary Conservation of Functionally Important Regions in H1N1 Influenza Proteome. PLoS ONE 2013, 8, e81027. [Google Scholar] [CrossRef]
- Wang, L.; Valderramos, S.G.; Wu, A.; Ouyang, S.; Li, C.; Brasil, P.; Bonaldo, M.; Coates, T.; Nielsen-Saines, K.; Jiang, T.; et al. From Mosquitos to Humans: Genetic Evolution of Zika Virus. Cell Host Microbe 2016, 19, 561–565. [Google Scholar] [CrossRef]
- Patel, H.; Kukol, A. Prediction of ligands to universally conserved binding sites of the influenza a virus nuclear export protein. Virology 2019, 537, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Brister, R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2014, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2017, 27, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; A Marti-Renom, M.; Madhusudhan, M.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank, in Protein Structure; CRC Press: Boca Raton, FL, USA, 2003; pp. 394–410. [Google Scholar]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Dong, N.; Yang, X.; Ye, L.; Chen, K.; Chan, E.W.-C.; Yang, M.; Chen, S. Genomic and protein structure modelling analysis depicts the origin and infectivity of SARS-CoV-2, a new coronavirus which caused a pneumonia outbreak in Wuhan, China. bioRxiv 2020. [Google Scholar] [CrossRef]
- De Chassey, B.; Meyniel-Schicklin, L.; Vonderscher, J.; André, P.; Lotteau, V. Virus-host interactomics: New insights and opportunities for antiviral drug discovery. Genome Med. 2014, 6, 115. [Google Scholar] [CrossRef]
- Von Brunn, A.; Teepe, C.; Simpson, J.C.; Pepperkok, R.; Friedel, C.C.; Zimmer, R.; Roberts, R.; Baric, R.; Haas, J. Analysis of Intraviral Protein-Protein Interactions of the SARS Coronavirus ORFeome. PLoS ONE 2007, 2, e459. [Google Scholar] [CrossRef]
- Pfefferle, S.; Schöpf, J.; Kögl, M.; Friedel, C.C.; Müller, M.A.; Carbajo-Lozoya, J.; Stellberger, T.; Von Dall’Armi, E.; Herzog, P.; Kallies, S.; et al. The SARS-Coronavirus-Host Interactome: Identification of Cyclophilins as Target for Pan-Coronavirus Inhibitors. PLoS Pathog. 2011, 7, e1002331. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscae: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A contemporary view of coronavirus transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Saikatendu, K.S.; Joseph, J.S.; Subramanian, V.; Clayton, T.; Griffith, M.; Moy, K.; Velasquez, J.; Neuman, B.W.; Buchmeier, M.J.; Stevens, R.C.; et al. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1 ″-phosphate dephosphorylation by a conserved domain of nsP3. Structure 2005, 13, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Serrano, P.; Johnson, M.A.; Almeida, M.S.; Horst, R.; Herrmann, T.; Joseph, J.S.; Neuman, B.W.; Subramanian, V.; Saikatendu, K.S.; Buchmeier, M.J.; et al. Nuclear magnetic resonance structure of the N-terminal domain of non-structural protein 3 from the severe acute respiratory syndrome coronavirus. J. Virol. 2007, 81, 12049–12060. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Cheng, S.; Brooks, C.L., III. Viral Capsid Proteins Are Segregated in Structural Fold Space. PLoS Comput. Boil. 2013, 9, e1002905. [Google Scholar] [CrossRef]
- Patel, H.; Kukol, A. Evolutionary conservation of influenza A PB2 sequences reveals potential target sites for small molecule inhibitors. Virology 2017, 509, 112–120. [Google Scholar] [CrossRef]
- Zhu, Z.; Chakraborti, S.; He, Y.; Roberts, A.; Sheahan, T.; Xiao, X.; Hensley, L.E.; Prabakaran, P.; Rockx, B.; Sidorov, I.; et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2007, 104, 12123–12128. [Google Scholar] [CrossRef]
- Sui, J.; Deming, M.; Rockx, B.; Liddington, R.C.; Zhu, Q.K.; Baric, R.S.; Marasco, W.A. Effects of Human Anti-Spike Protein Receptor Binding Domain Antibodies on Severe Acute Respiratory Syndrome Coronavirus Neutralization Escape and Fitness. J. Virol. 2014, 88, 13769–13780. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, M.M.; Babcook, J.; Prabhakar, B.S. Human monoclonal antibodies to SARS-coronavirus inhibit infection by different mechanisms. Virology 2009, 394, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; E Gorbalenya, A.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.J.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Krüger, N.; Müller, M.; Drosten, C.; Pöhlmann, S.; Müller, M.A. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kruse, R.L. Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000 Res. 2020, 9, 72. [Google Scholar] [CrossRef]
- Voitenko, O.S.; Dhroso, A.; Feldmann, A.; Korkin, D.; Kalinina, O.V. Patterns of amino acid conservation in human and animal immunodeficiency viruses. Bioinformatics 2016, 32, i685–i692. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Accession | wORF1ab Region | Modeled Length | Template PDB id | Trgt-Tmplt Seq ID | Organism |
|---|---|---|---|---|---|---|
| wS, surface glycoprotein | YP_009724390 | 1273 | 6ACK | 75% | SARS-CoV | |
| wE, envelope protein | YP_009724392 | 75 | 5X29 | 89% | SARS-CoV | |
| wORF7a | YP_009724395 | 121 | 1YO4 | 90% | SARS-CoV | |
| wN, nucleocapsid phosphoprotein | YP_009724397 | 419 | 2JW8 | 96% | SARS-CoV | |
| 1SSK | 83% | SARS-CoV | ||||
| 4UD1 | 51% | MERS-CoV | ||||
| wNsp1 | YP_009725297 | 13-127 | 115 | 2HSX | 86% | SARS-CoV |
| wNsp3-domain1 | YP_009725299 | 819-926 | 107 | 2GRI | 79% | SARS-CoV |
| wNsp3-domain2 | YP_009725299 | 1024-1198 | 175 | 2ACF | 72% | SARS-CoV |
| wNsp3-domain3 | YP_009725299 | 1232-1494 | 263 | 2WCT | 76% | SARS-CoV |
| wNsp3-domain4 | YP_009725299 | 1495-1550 | 66 | 2KAF | 70% | SARS-CoV |
| wNsp3-domain5 | YP_009725299 | 1564-1878 | 315 | 3E9S | 82% | SARS-CoV |
| wNsp3-domain6 | YP_009725299 | 1908-2763 | 113 | 2K87 | 82% | SARS-CoV |
| wNsp4 | YP_009725300 | 3173-3263 | 91 | 3VC8 | 60% | MHV |
| wNsp5 | YP_009725301 | 3264-3569 | 306 | 2GT7 | 96% | SARS-CoV |
| wNsp7 | YP_009725302 | 3860-3942 | 83 | 1YSY | 67% | SARS-CoV |
| wNsp8 | YP_009725304 | 4019-4132 | 114 | 6NUR | 85% | SARS-CoV |
| wNsp9 | YP_009725305 | 4041-4253 | 113 | 3EE7 | 99% | SARS-CoV |
| wNsp10 | YP_009725306 | 4262-4382 | 121 | 2G9T | 98% | SARS-CoV |
| wNsp12 | YP_009725307 | 4542-5311 | 770 | 6NUR | 97% | SARS-CoV |
| wNsp13 | YP_009725308 | 5325-5920 | 596 | 6JYT | 100% | SARS-CoV |
| wNsp14 | YP_009725309 | 5926-6451 | 526 | 5C8U | 95% | SARS-CoV |
| wNsp15 | YP_009725310 | 6452-6797 | 346 | 2H85 | 86% | SARS-CoV |
| wNsp16 | YP_009725311 | 6800-7087 | 288 | 2XYQ | 94% | SARS-CoV |
| Network Parameters | SARS-CoV Intra-viral Interactome | SARS-CoV-Host Interactome | Unified Interactome |
|---|---|---|---|
| No. of nodes | 31 | 118 | 125 |
| No. of edges | 86 | 114 | 206 |
| No. of components | 1 | 8 | 2 |
| Diameter | 4 | 14 | 7 |
| Average degree | 4.710 | 1.95 | 3.04 |
| Clustering coefficient | 0.448 | 0.0 | 0.068 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses 2020, 12, 360. https://doi.org/10.3390/v12040360
Srinivasan S, Cui H, Gao Z, Liu M, Lu S, Mkandawire W, Narykov O, Sun M, Korkin D. Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses. 2020; 12(4):360. https://doi.org/10.3390/v12040360
Chicago/Turabian StyleSrinivasan, Suhas, Hongzhu Cui, Ziyang Gao, Ming Liu, Senbao Lu, Winnie Mkandawire, Oleksandr Narykov, Mo Sun, and Dmitry Korkin. 2020. "Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins" Viruses 12, no. 4: 360. https://doi.org/10.3390/v12040360
APA StyleSrinivasan, S., Cui, H., Gao, Z., Liu, M., Lu, S., Mkandawire, W., Narykov, O., Sun, M., & Korkin, D. (2020). Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses, 12(4), 360. https://doi.org/10.3390/v12040360