Structural and Functional Characterization of the Secondary Mutation N126K Selected by Various HIV-1 Fusion Inhibitors

 ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Cells and Plasmids
2.2. Peptide Synthesis
2.3. Site-Directed Mutagenesis
2.4. Single-Cycle Infection Assay
2.5. Cell-Cell Fusion Assay
2.6. Capture ELISA
2.7. Western Blotting Assay
2.8. Flow Cytometry Assay
2.9. Circular Dichroism (CD) Spectroscopy
2.10. Isothermal Titration Calorimetry (ITC)
2.11. Crystallization and Structure Determination
3. Results
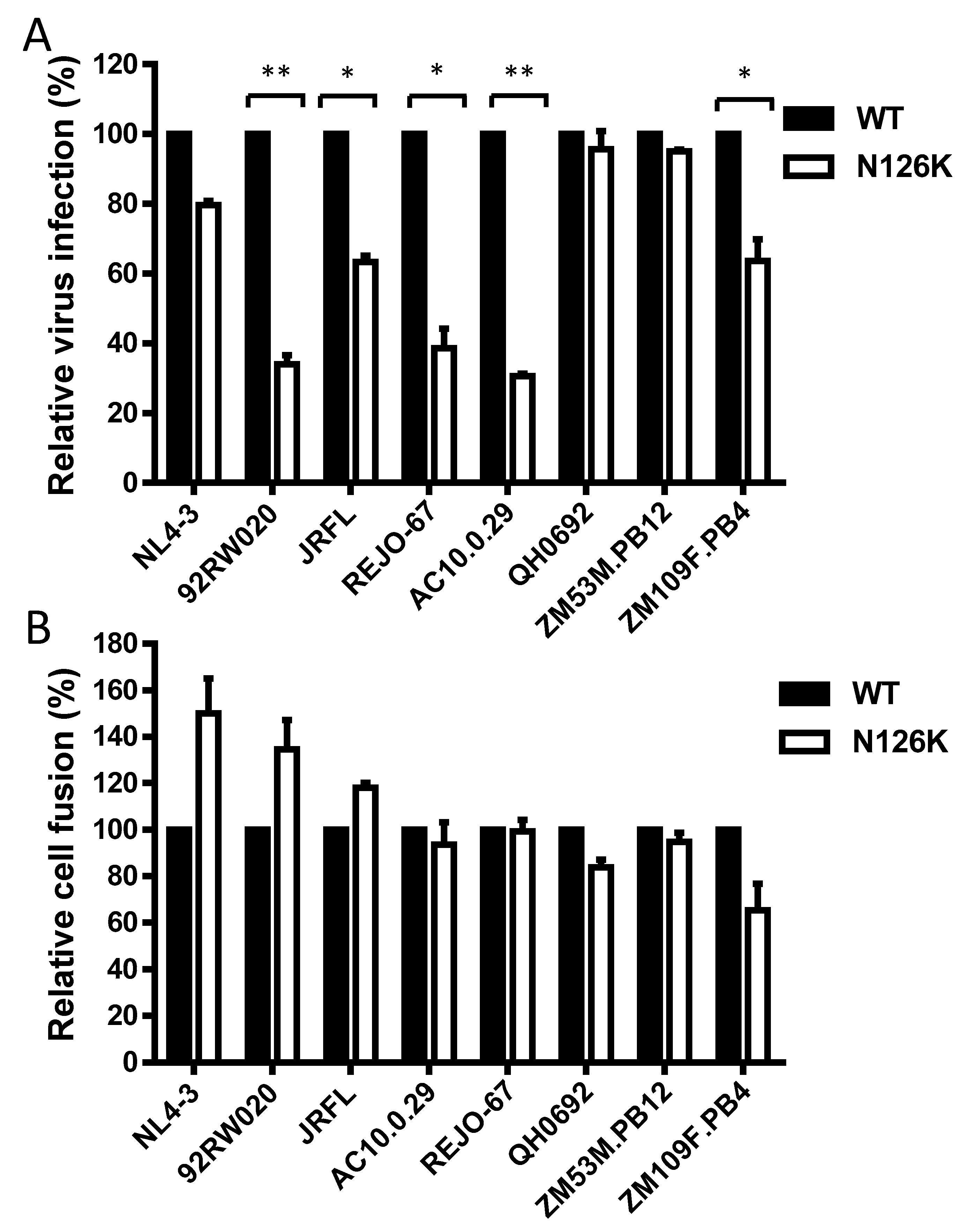
3.1. Single N126K Mutation-Mediated Resistance Profiles in Divergent HIV-1 Isolates
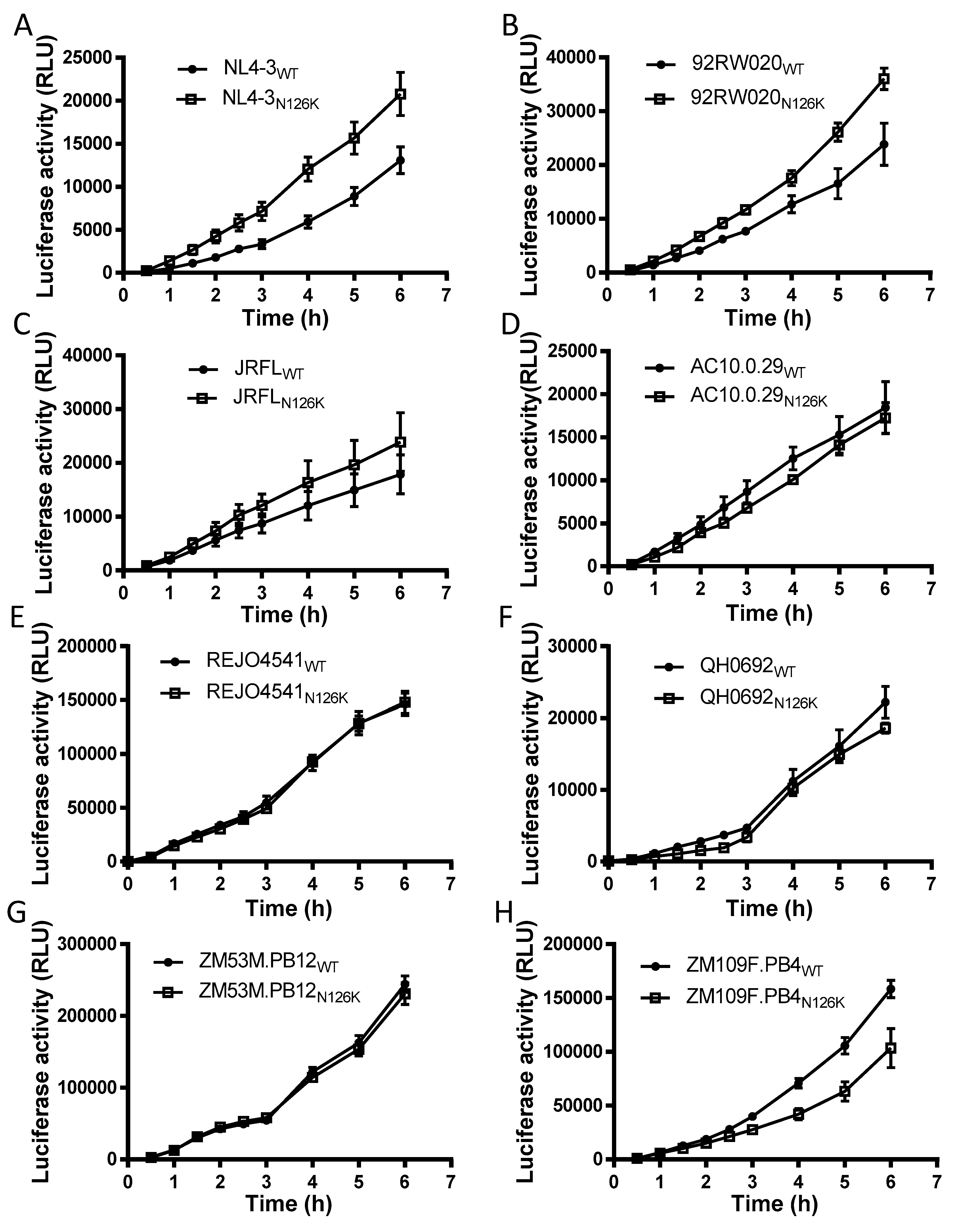
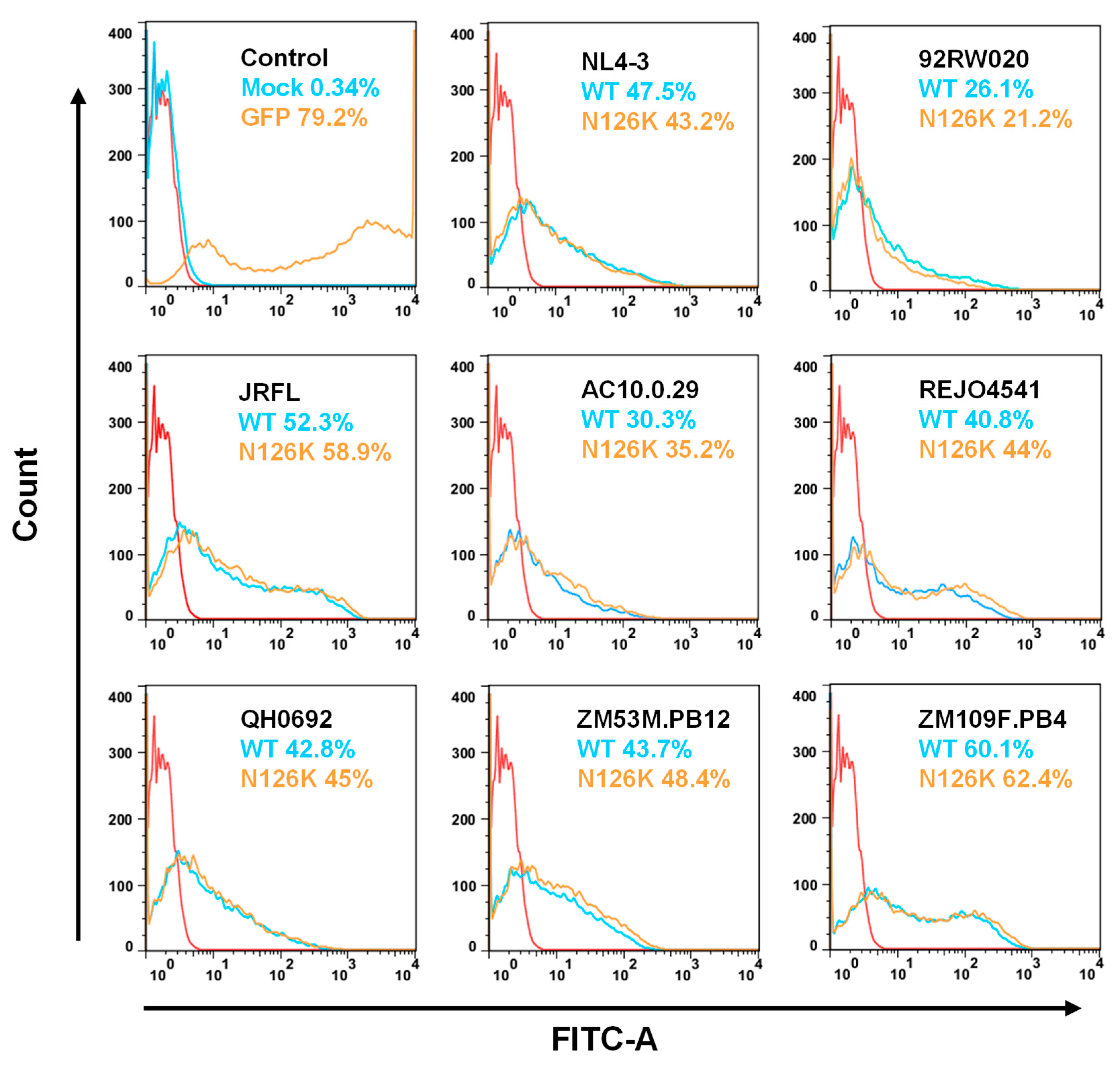
3.2. Effect of N126 Mutation on the Functionality of Divergent HIV-1 Envs
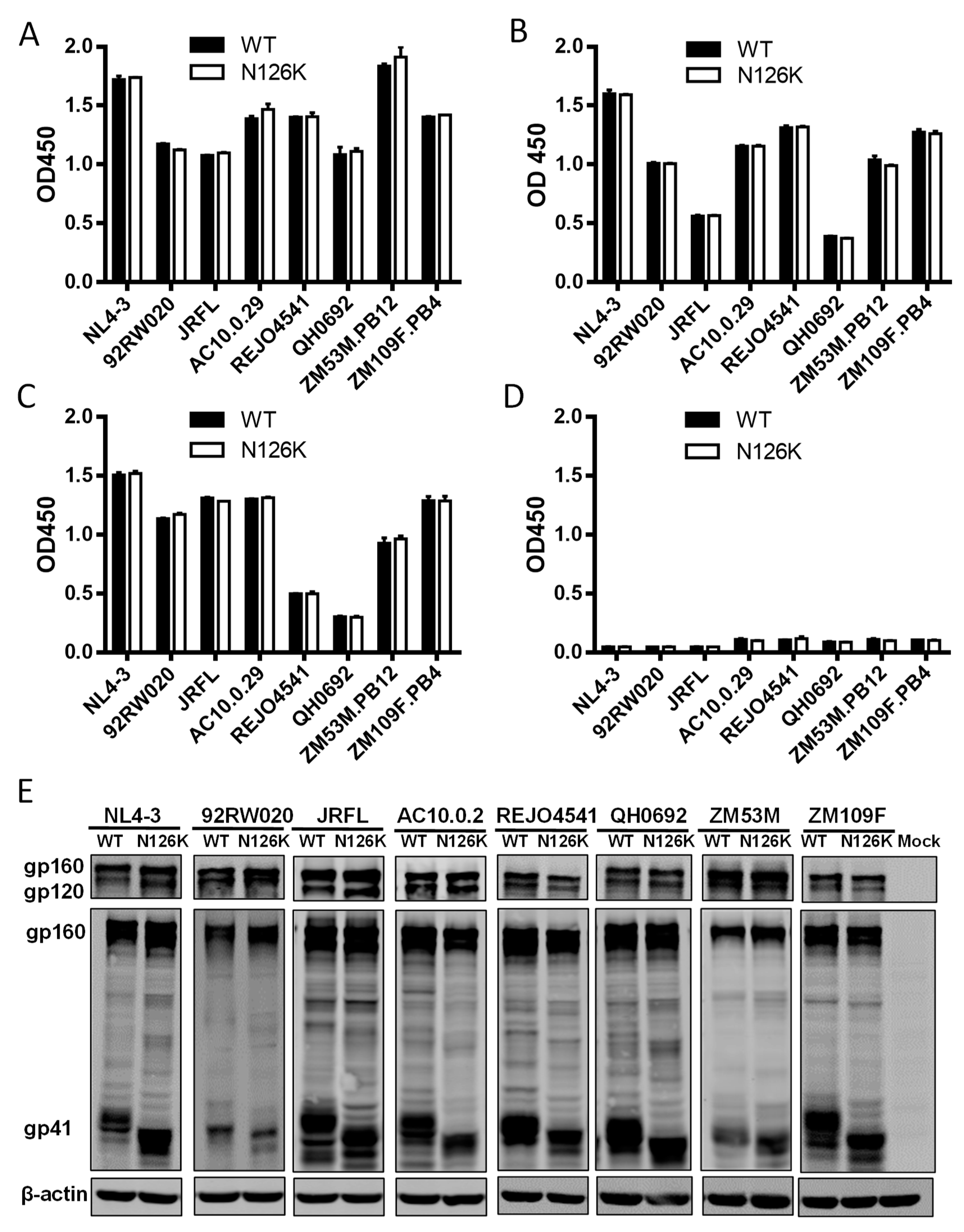
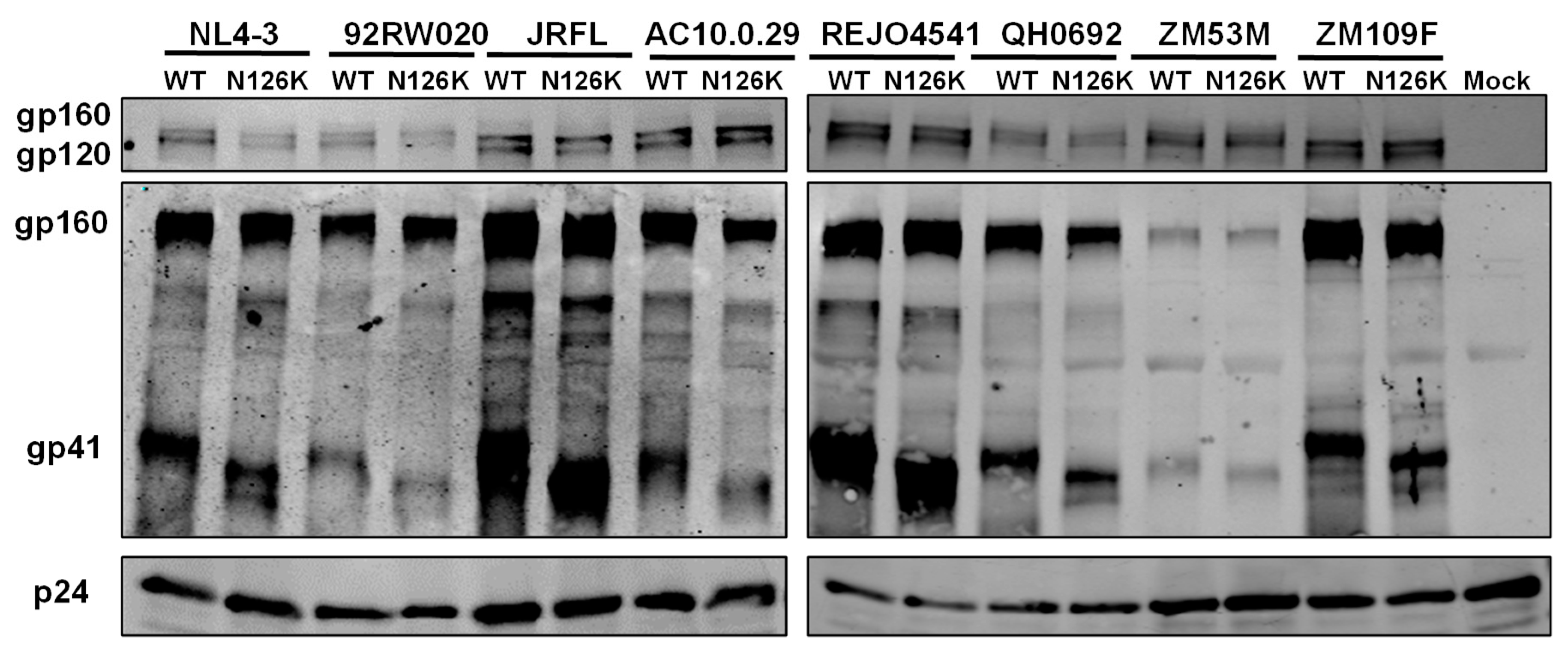
3.3. N126K Mutation Does Not Affect the Expression of Viral Env Glycoprotein but Causes Deglycosylation in Gp41
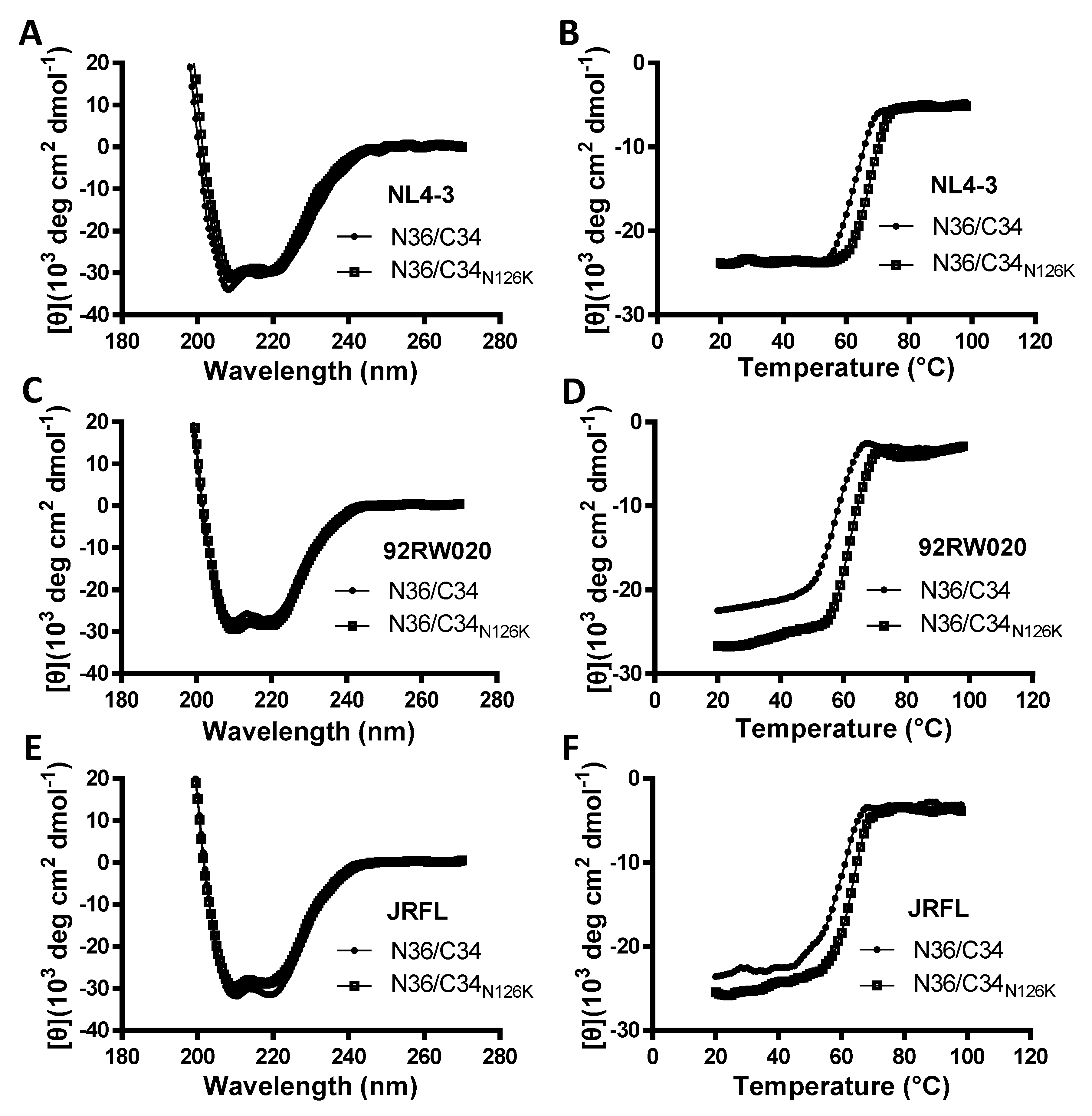
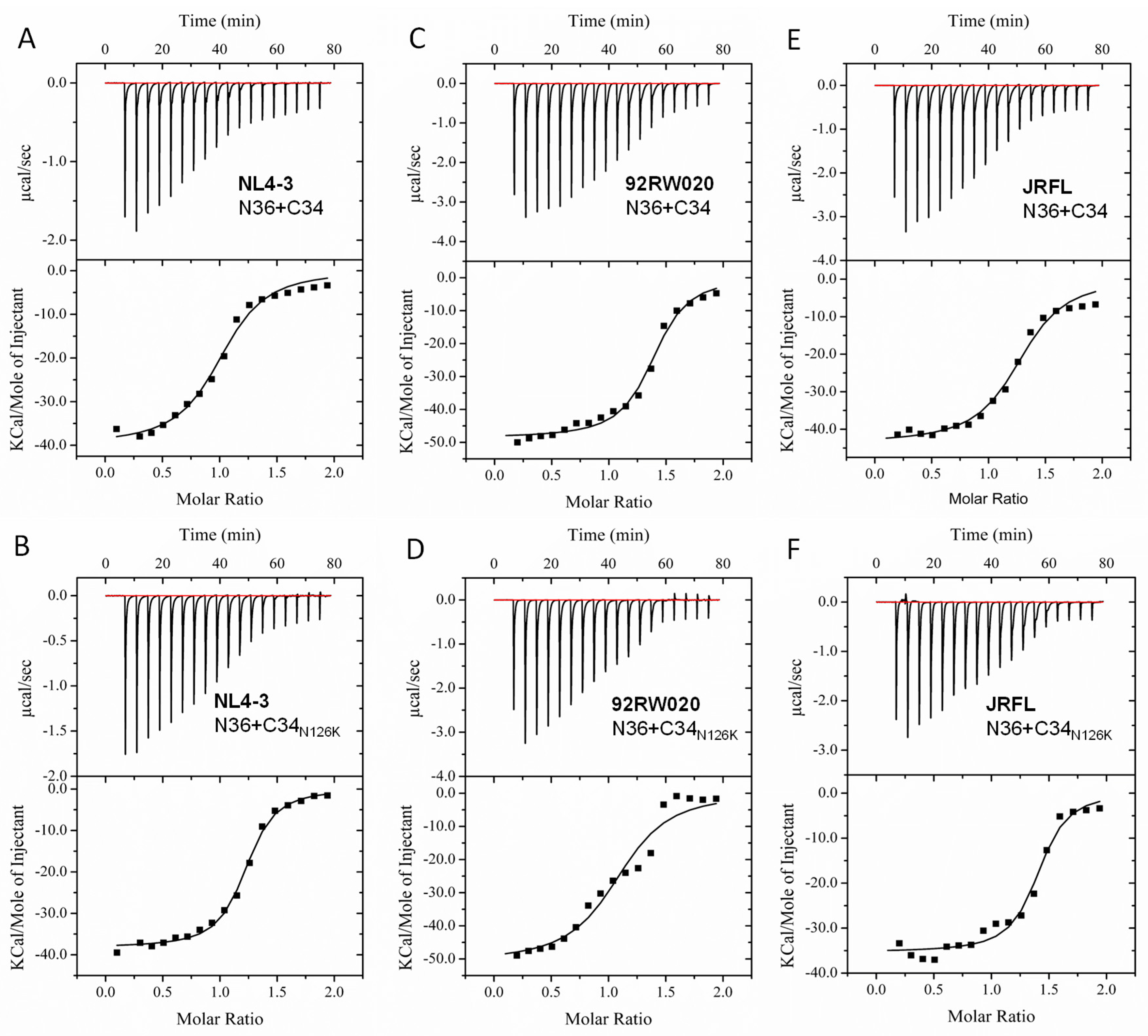
3.4. N126K Mutation Can Significantly Increases the Thermostability of 6-HB Structure
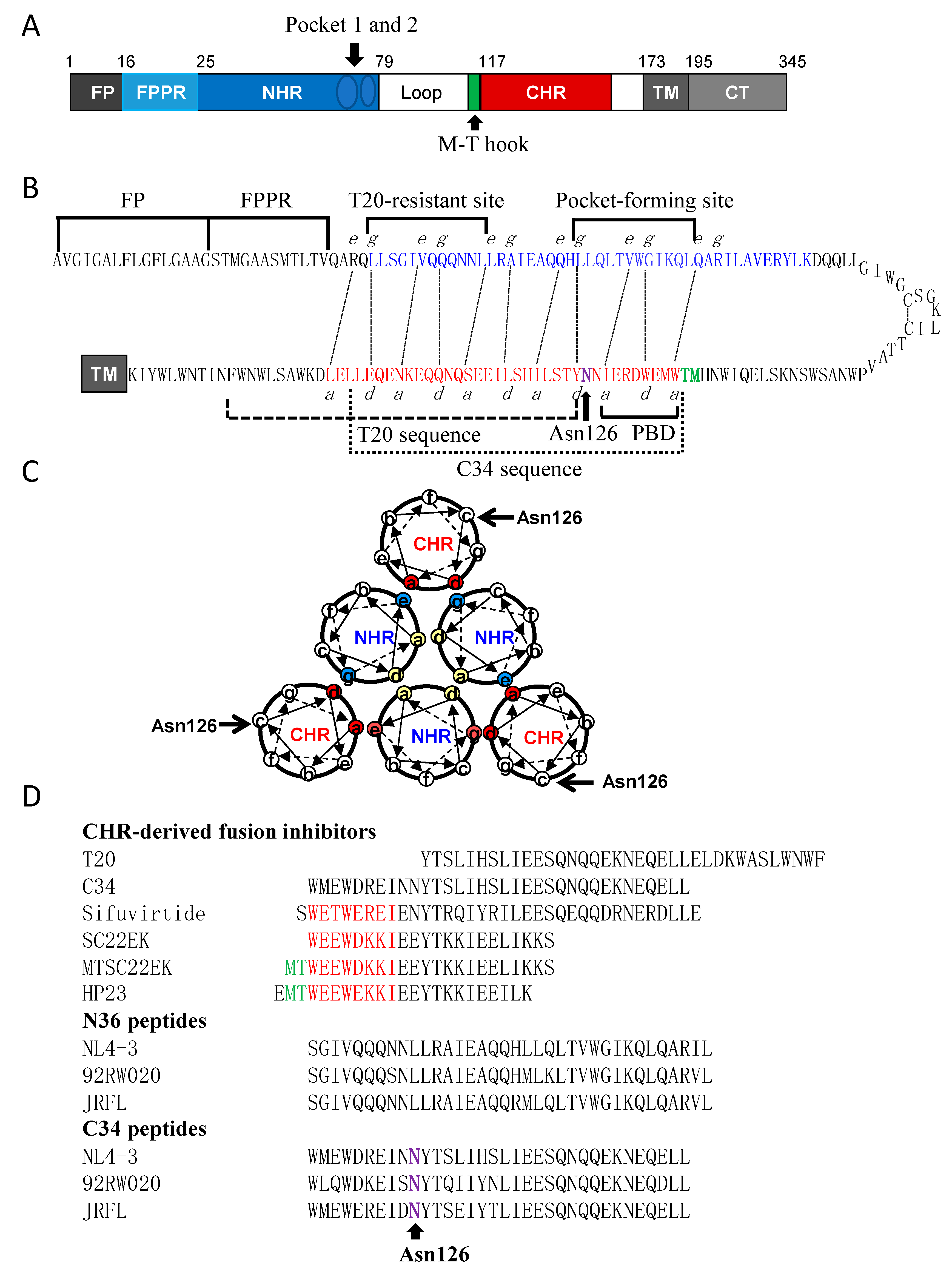
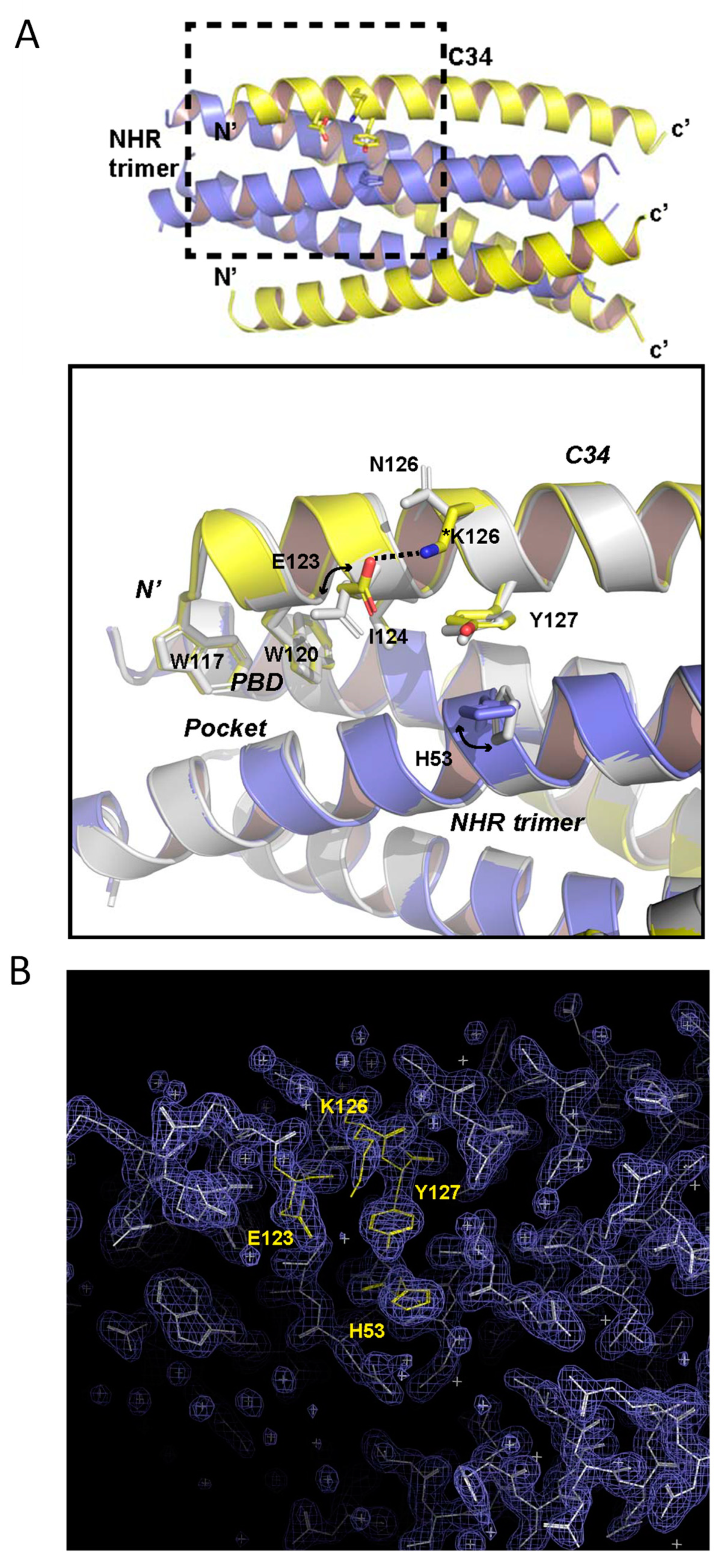
3.5. Structural Basis of N126K Mutation in 6-HB
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Eckert, D.M.; Kim, P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001, 70, 777–810. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Qiu, Z.; Chong, H.; Yao, X.; Su, Y.; Cui, S.; He, Y. Identification and characterization of a subpocket on the N-trimer of HIV-1 Gp41: Implication for viral entry and drug target. Aids 2015, 29, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Chutkowski, C.T.; Kim, P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 1998, 95, 15613–15617. [Google Scholar] [CrossRef]
- Crespillo, S.; Camara-Artigas, A.; Casares, S.; Morel, B.; Cobos, E.S.; Mateo, P.L.; Mouz, N.; Martin, C.E.; Roger, M.G.; El Habib, R.; et al. Single-chain protein mimetics of the N-terminal heptad-repeat region of gp41 with potential as anti-HIV-1 drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 18207–18212. [Google Scholar] [CrossRef]
- Chu, S.; Gochin, M. Identification of fragments targeting an alternative pocket on HIV-1 gp41 by NMR screening and similarity searching. Bioorganic Med. Chem. Lett. 2013, 23, 5114–5118. [Google Scholar] [CrossRef]
- He, Y. Synthesized peptide inhibitors of HIV-1 gp41-dependent membrane fusion. Curr. Pharm. Des. 2013, 19, 1800–1809. [Google Scholar] [CrossRef]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998, 72, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.E.; Sanders, R.W.; Deng, Y.; Jurriaans, S.; Lange, J.M.; Lu, M.; Berkhout, B. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J. Virol. 2004, 78, 12428–12437. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Wang, Q.; Xu, W.; Lu, L.; Jiang, S. Development of Protein- and Peptide-Based HIV Entry Inhibitors Targeting gp120 or gp41. Viruses 2019, 11, 705. [Google Scholar] [CrossRef]
- Otaka, A.; Nakamura, M.; Nameki, D.; Kodama, E.; Uchiyama, S.; Nakamura, S.; Nakano, H.; Tamamura, H.; Kobayashi, Y.; Matsuoka, M.; et al. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew. Chem. 2002, 41, 2937–2940. [Google Scholar] [CrossRef]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar] [CrossRef]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA 2007, 104, 12772–12777. [Google Scholar] [CrossRef]
- Xiong, S.; Borrego, P.; Ding, X.; Zhu, Y.; Martins, A.; Chong, H.; Taveira, N.; He, Y. A Helical Short-Peptide Fusion Inhibitor with Highly Potent Activity against Human Immunodeficiency Virus Type 1 (HIV-1), HIV-2, and Simian Immunodeficiency Virus. J. Virol. 2017, 91, e01839-16. [Google Scholar] [CrossRef]
- Chong, H.; Qiu, Z.; Su, Y.; Yang, L.; He, Y. Design of a highly potent HIV-1 fusion inhibitor targeting the gp41 pocket. Aids 2015, 29, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Yao, X.; Qiu, Z.; Sun, J.; Zhang, M.; Waltersperger, S.; Wang, M.; Liu, S.L.; Cui, S.; He, Y. Short-peptide fusion inhibitors with high potency against wild-type and enfuvirtide-resistant HIV-1. FASEB J. 2013, 27, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chong, H.; Xiong, S.; Qiao, Y.; Qiu, Z.; He, Y. Genetic Pathway of HIV-1 Resistance to Novel Fusion Inhibitors Targeting the Gp41 Pocket. J. Virol. 2015, 89, 12467–12479. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chong, H.; Qiu, Z.; Xiong, S.; He, Y. Mechanism of HIV-1 Resistance to Short-Peptide Fusion Inhibitors Targeting the Gp41 Pocket. J. Virol. 2015, 89, 5801–5811. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Ding, X.; Liu, Z.; Wu, X.; Zhu, Y.; Wei, H.; Chong, H.; Cui, S.; He, Y. Molecular mechanism of HIV-1 resistance to sifuvirtide, a clinical trial-approved membrane fusion inhibitor. J. Biol. Chem. 2018, 293, 12703–12718. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Z.; Ding, X.; Yu, D.; Wei, H.; Qin, B.; Zhu, Y.; Chong, H.; Cui, S.; He, Y. Mechanism of HIV-1 Resistance to an Electronically Constrained alpha-Helical Peptide Membrane Fusion Inhibitor. J. Virol. 2018, 92, e02044-17. [Google Scholar] [CrossRef] [PubMed]
- Eggink, D.; Baldwin, C.E.; Deng, Y.; Langedijk, J.P.; Lu, M.; Sanders, R.W.; Berkhout, B. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J. Virol. 2008, 82, 6678–6688. [Google Scholar] [CrossRef]
- Eggink, D.; Bontjer, I.; Langedijk, J.P.; Berkhout, B.; Sanders, R.W. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J. Virol. 2011, 85, 10785–10797. [Google Scholar] [CrossRef]
- Shimura, K.; Nameki, D.; Kajiwara, K.; Watanabe, K.; Sakagami, Y.; Oishi, S.; Fujii, N.; Matsuoka, M.; Sarafianos, S.G.; Kodama, E.N. Resistance profiles of novel electrostatically constrained HIV-1 fusion inhibitors. J. Biol. Chem. 2010, 285, 39471–39480. [Google Scholar] [CrossRef]
- Nameki, D.; Kodama, E.; Ikeuchi, M.; Mabuchi, N.; Otaka, A.; Tamamura, H.; Ohno, M.; Fujii, N.; Matsuoka, M. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and Rev-responsive element functions. J. Virol. 2005, 79, 764–770. [Google Scholar] [CrossRef]
- Geng, X.; Liu, Z.; Yu, D.; Qin, B.; Zhu, Y.; Cui, S.; Chong, H.; He, Y. Conserved Residue Asn-145 in the C-Terminal Heptad Repeat Region of HIV-1 gp41 is Critical for Viral Fusion and Regulates the Antiviral Activity of Fusion Inhibitors. Viruses 2019, 11, 609. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ding, X.; Yu, D.; Chong, H.; He, Y. The Tryptophan-Rich Motif of HIV-1 Gp41 Can Interact with the N-Terminal Deep Pocket Site: New Insights into the Structure and Function of Gp41 and Its Inhibitors. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lohrengel, S.; Hermann, F.; Hagmann, I.; Oberwinkler, H.; Scrivano, L.; Hoffmann, C.; von Laer, D.; Dittmar, M.T. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J. Virol. 2005, 79, 10237–10246. [Google Scholar] [CrossRef] [PubMed]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. Aids 2004, 18, 1787–1794. [Google Scholar] [CrossRef]
- Liu, Z.; Shan, M.; Li, L.; Lu, L.; Meng, S.; Chen, C.; He, Y.; Jiang, S.; Zhang, L. In vitro selection and characterization of HIV-1 variants with increased resistance to sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2011, 286, 3277–3287. [Google Scholar] [CrossRef]
- Cabrera, C.; Marfil, S.; Garcia, E.; Martinez-Picado, J.; Bonjoch, A.; Bofill, M.; Moreno, S.; Ribera, E.; Domingo, P.; Clotet, B.; et al. Genetic evolution of gp41 reveals a highly exclusive relationship between codons 36, 38 and 43 in gp41 under long-term enfuvirtide-containing salvage regimen. Aids 2006, 20, 2075–2080. [Google Scholar] [CrossRef]
- Xu, L.; Pozniak, A.; Wildfire, A.; Stanfield-Oakley, S.A.; Mosier, S.M.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar] [CrossRef]
- Loutfy, M.R.; Raboud, J.M.; Montaner, J.S.; Antoniou, T.; Wynhoven, B.; Smaill, F.; Rouleau, D.; Gill, J.; Schlech, W.; Brumme, Z.L.; et al. Assay of HIV gp41 amino acid sequence to identify baseline variation and mutation development in patients with virologic failure on enfuvirtide. Antivir. Res. 2007, 75, 58–63. [Google Scholar] [CrossRef]
- Poveda, E.; Rodes, B.; Labernardiere, J.L.; Benito, J.M.; Toro, C.; Gonzalez-Lahoz, J.; Faudon, J.L.; Clavel, F.; Schapiro, J.; Soriano, V. Evolution of genotypic and phenotypic resistance to Enfuvirtide in HIV-infected patients experiencing prolonged virologic failure. J. Med. Virol. 2004, 74, 21–28. [Google Scholar] [CrossRef]
- Ray, N.; Blackburn, L.A.; Doms, R.W. HR-2 mutations in human immunodeficiency virus type 1 gp41 restore fusion kinetics delayed by HR-1 mutations that cause clinical resistance to enfuvirtide. J. Virol. 2009, 83, 2989–2995. [Google Scholar] [CrossRef]
- Ray, N.; Harrison, J.E.; Blackburn, L.A.; Martin, J.N.; Deeks, S.G.; Doms, R.W. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J. Virol. 2007, 81, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Svicher, V.; Aquaro, S.; D’Arrigo, R.; Artese, A.; Dimonte, S.; Alcaro, S.; Santoro, M.M.; Di Perri, G.; Caputo, S.L.; Bellagamba, R.; et al. Specific enfuvirtide-associated mutational pathways in HIV-1 Gp41 are significantly correlated with an increase in CD4(+) cell count, despite virological failure. J. Infect. Dis. 2008, 197, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Eggink, D.; Langedijk, J.P.; Bonvin, A.M.; Deng, Y.; Lu, M.; Berkhout, B.; Sanders, R.W. Detailed mechanistic insights into HIV-1 sensitivity to three generations of fusion inhibitors. J. Biol. Chem. 2009, 284, 26941–26950. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Chong, H.; Zhang, C.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J. Biol. Chem. 2012, 287, 6788–6796. [Google Scholar] [CrossRef]
- Sivaraman, V.; Zhang, L.; Meissner, E.G.; Jeffrey, J.L.; Su, L. The heptad repeat 2 domain is a major determinant for enhanced human immunodeficiency virus type 1 (HIV-1) fusion and pathogenicity of a highly pathogenic HIV-1 Env. J. Virol. 2009, 83, 11715–11725. [Google Scholar] [CrossRef]
- Izumi, K.; Kodama, E.; Shimura, K.; Sakagami, Y.; Watanabe, K.; Ito, S.; Watabe, T.; Terakawa, Y.; Nishikawa, H.; Sarafianos, S.G.; et al. Design of peptide-based inhibitors for human immunodeficiency virus type 1 strains resistant to T-20. J. Biol. Chem. 2009, 284, 4914–4920. [Google Scholar] [CrossRef]
- Desmezieres, E.; Gupta, N.; Vassell, R.; He, Y.; Peden, K.; Sirota, L.; Yang, Z.; Wingfield, P.; Weiss, C.D. Human immunodeficiency virus (HIV) gp41 escape mutants: Cross-resistance to peptide inhibitors of HIV fusion and altered receptor activation of gp120. J. Virol. 2005, 79, 4774–4781. [Google Scholar] [CrossRef]
- De Feo, C.J.; Wang, W.; Hsieh, M.L.; Zhuang, M.; Vassell, R.; Weiss, C.D. Resistance to N-peptide fusion inhibitors correlates with thermodynamic stability of the gp41 six-helix bundle but not HIV entry kinetics. Retrovirology 2014, 11, 86. [Google Scholar] [CrossRef]
- Chong, H.; Yao, X.; Qiu, Z.; Sun, J.; Qiao, Y.; Zhang, M.; Wang, M.; Cui, S.; He, Y. The M-T hook structure increases the potency of HIV-1 fusion inhibitor sifuvirtide and overcomes drug resistance. J. Antimicrob. Chemother. 2014, 69, 6759. [Google Scholar] [CrossRef]
- Chong, H.; Qiu, Z.; Sun, J.; Qiao, Y.; Li, X.; He, Y. Two M-T hook residues greatly improve the antiviral activity and resistance profile of the HIV-1 fusion inhibitor SC29EK. Retrovirology 2014, 11, 40. [Google Scholar] [CrossRef]
- Wang, L.X.; Song, H.; Liu, S.; Lu, H.; Jiang, S.; Ni, J.; Li, H. Chemoenzymatic synthesis of HIV-1 gp41 glycopeptides: Effects of glycosylation on the anti-HIV activity and alpha-helix bundle-forming ability of peptide C34. Chembiochem 2005, 6, 1068–1074. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | N36/C34N126K |
|---|---|
| Data collection | |
| Space group | C121 |
| Cell dimensions | |
| a,b,c | 88.86 Å 50.81 Å 56.11 Å |
| α,β,γ | 90.00° 90.40° 90.00° |
| X-ray source | SSRF BEAMLINE BL19U1 |
| Wavelength | 0.98 Å |
| Data range | 50–1.65 (1.75–1.65) Å |
| Reflections unique | 29,914 |
| Rmerge (%) | 5.3 (83.3) |
| I/σI | 17.79 (1.94) |
| Completeness (last shell) | 98.9% (97.9%) |
| Redundancy (last shell) | 6.54 (6.23) |
| Refinement | |
| Resolution range | 44.11–1.65 (1.70–1.65) Å |
| No.Reflections | 29,894 |
| R work a/R free b | 0.2062/0.2258 |
| Nonhydrogen atoms | 1987 |
| Protein | 1785 |
| Water | 202 |
| N-Acetyl group | 6 |
| B-Factor averages | 34.0 Å2 |
| Root mean square deviation | |
| Bond length | 0.025 Å |
| Bond angles | 1.35° |
| Validation | |
| MolProbity score | 1.49, rating 91st percentile among structures of comparable resolution |
| % Favored regions and Outliers in Ramachandran plot | 100.0, 0.0, 0.0 |
| HIV-1 | Subtype | T20 | C34 | SFT | SC22EK | MTSC22EK | HP23 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | FC | IC50 (nM) | FC | IC50 (nM) | FC | IC50 (nM) | FC | IC50 (nM) | FC | IC50 (nM) | FC | ||
| NL4-3WT | B | 71± 13.8 | 1.5 ± 0.4 | 1.9 ± 0.8 | 47.9 ± 12.9 | 2 ± 0.8 | 0.6 ± 0.2 | ||||||
| NL4-3N126K | B | 153.4 ± 2.2 | 2.2 | 5.7 ± 2 | 3.8 | 5.1 ± 1 | 2.7 | 86.5 ± 1.8 | 1.8 | 4.9 ± 1.6 | 2.5 | 1.5 ± 0.2 | 2.6 |
| 92RW020WT | A | 16.3 ± 6.2 | 4.8 ± 0.5 | 7.5 ± 3.1 | 427.7 ± 56.9 | 6.7 ± 2.8 | 2.1 ± 1 | ||||||
| 92RW020N126K | A | 126.8 ± 124.1 | 7.8 | 6.2 ± 0.9 | 1.3 | 10.9 ± 1.3 | 1.5 | 1333.5 ± 766.2 | 3.1 | 14.3 ± 6.8 | 2.1 | 3.3 ± 1 | 1.5 |
| JRFLWT | B | 13.9 ± 2.1 | 5.8 ± 2.4 | 9 ± 2.1 | 467.2 ± 104.6 | 19.5 ± 6.6 | 5.9 ± 2 | ||||||
| JRFLN126K | B | 137.4 ± 52.6 | 9.9 | 27.9 ± 16.5 | 4.8 | 31.3 ± 12.9 | 3.5 | 600.4 ± 93.3 | 1.3 | 57.4 ± 18.4 | 3 | 15.3 ± 0.2 | 2.6 |
| REJO4541WT | B | 19 ± 1.7 | 9.9 ± 4.4 | 6.1 ± 0.2 | 381.7 ± 95.5 | 9.6 ± 2.5 | 3.1 ± 0.5 | ||||||
| REJO4541N126K | B | 66.9 ± 14 | 3.5 | 15 ± 13.8 | 1.5 | 10.5 ± 1.9 | 1.7 | 1284 ± 78.2 | 3.4 | 13.2 ± 3.8 | 1.4 | 5.7 ± 2 | 1.8 |
| AC10.29WT | B | 13.2 ± 7.1 | 1.4 ± 0.2 | 2.1 ± 1 | 110.9 ± 37.1 | 3.1 ± 0.7 | 0.7 ± 0.3 | ||||||
| AC10.29N126K | B | 36.4 ± 1.4 | 2.8 | 1.8 ± 0.3 | 1.3 | 2.5 ± 0.9 | 1.2 | 247.7 ± 38.7 | 2.2 | 4.1 ± 1.6 | 1.3 | 1.2 ± 0 | 1.6 |
| QH0692WT | B | 92.1 ± 19.9 | 59.1 ± 8.4 | 28.6 ± 1.5 | 2055 ± 291.9 | 68.9 ± 7.5 | 34.6 ± 4.9 | ||||||
| QH0692N126K | B | 184.9 ± 44.5 | 2 | 32.3 ± 4.9 | 0.6 | 62.5 ± 7.7 | 2.2 | 1163.3 ± 202.7 | 0.6 | 92.2 ± 22.4 | 1.3 | 41.9 ± 3 | 1.2 |
| ZM53M.PB12WT | C | 21.4 ± 10.8 | 10.7 ± 6.2 | 2.1 ± 0.8 | 47.3 ± 3.4 | 2.7 ± 0.2 | 1.2 ± 0.1 | ||||||
| ZM53M.PB12N126K | C | 49.8 ± 7.8 | 2.3 | 15.7 ± 3.3 | 1.5 | 2.8 ± 0.9 | 1.3 | 164.5 ± 18.7 | 3.5 | 3.2 ± 0.7 | 1.2 | 1.6 ± 0.3 | 1.3 |
| ZM109F.PB4WT | C | 12.9 ± 3.5 | 1.5 ± 0.3 | 2.6 ± 1.1 | 43.6 ± 8.1 | 2 ± 0.4 | 0.8 ± 0.1 | ||||||
| ZM109F.PB4N126K | C | 23.8 ± 7.5 | 1.9 | 2.8 ± 0.5 | 1.8 | 4.5 ± 0.4 | 1.7 | 79.4 ± 6.9 | 1.8 | 3.3 ± 0.4 | 1.6 | 1.4 ± 0.3 | 1.8 |
| Peptide Pair | CD Data | ITC Data | ||||
|---|---|---|---|---|---|---|
| Helix (%) | Tm (°C) | N (Site) | K (M-1) | ΔH (cal/mol) | ΔS (cal/mol/deg) | |
| NL4-3 N36/C34 | 94.7 ± 3.7 | 63.8 ± 0.7 | 1 | 6.4 × 105 ± 1.2 × 105 | −4 × 104 ± 1059 | −112.5 ± 9.2 |
| NL4-3 N36/C34N126K | 91.2 ± 3.8 | 68.5 ± 0.8 | 1.2 | 1.9 × 106 ± 2.4 × 105 | −3.8 × 104 ± 427.5 | −94.5 ± 6.9 |
| 92RW020 N36/C34 | 79.1 ± 4.1 | 57.4 ± 0.8 | 1.4 | 9.4 × 105 ± 1.8 × 105 | −4.9 × 104 ± 787.7 | −121.6 ± 14.2 |
| 92RW020 N36/C34N126K | 78.8 ± 3.7 | 62.7 ± 0.6 | 1 | 1.3 × 106 ± 5 × 105 | −5.5 × 104 ± 1862 | −151.5 ± 20.4 |
| JRFL N36/C34 | 82.3 ± 6.1 | 57.9 ± 0.8 | 1.3 | 4.3 × 105 ± 8.7 × 104 | −4.4 × 104 ± 1005 | −123.5 ± 10 |
| JRFL N36/C34N126K | 82.8 ± 4.9 | 62.7 ± 0.7 | 1.4 | 1.2 × 106 ± 3.4 × 105 | −3.5 × 104 ± 748.6 | −92.9 ± 7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, D.; Su, Y.; Ding, X.; Zhu, Y.; Qin, B.; Chong, H.; Cui, S.; He, Y. Structural and Functional Characterization of the Secondary Mutation N126K Selected by Various HIV-1 Fusion Inhibitors. Viruses 2020, 12, 326. https://doi.org/10.3390/v12030326
Yu D, Su Y, Ding X, Zhu Y, Qin B, Chong H, Cui S, He Y. Structural and Functional Characterization of the Secondary Mutation N126K Selected by Various HIV-1 Fusion Inhibitors. Viruses. 2020; 12(3):326. https://doi.org/10.3390/v12030326
Chicago/Turabian StyleYu, Danwei, Yang Su, Xiaohui Ding, Yuanmei Zhu, Bo Qin, Huihui Chong, Sheng Cui, and Yuxian He. 2020. "Structural and Functional Characterization of the Secondary Mutation N126K Selected by Various HIV-1 Fusion Inhibitors" Viruses 12, no. 3: 326. https://doi.org/10.3390/v12030326
APA StyleYu, D., Su, Y., Ding, X., Zhu, Y., Qin, B., Chong, H., Cui, S., & He, Y. (2020). Structural and Functional Characterization of the Secondary Mutation N126K Selected by Various HIV-1 Fusion Inhibitors. Viruses, 12(3), 326. https://doi.org/10.3390/v12030326