La Crosse Virus Infection of Human Keratinocytes Leads to Interferon-Dependent Apoptosis of Bystander Non-Infected Cells In Vitro


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Infections
2.2. Western Blotting
2.3. Cell Viability and Caspase Assays
2.4. ZVAD Treatment
2.5. Supernatant Preparation
2.6. Reverse Transcription and Real-Time PCR
2.7. Interferon, Ruxolitinib and Neutralizing Antibody Treatment
2.8. Cell Viability and Virus Infection Quantification
2.9. Statistical Analyses
3. Results
3.1. HaCaT Cells Are Permissive to LACV Infection
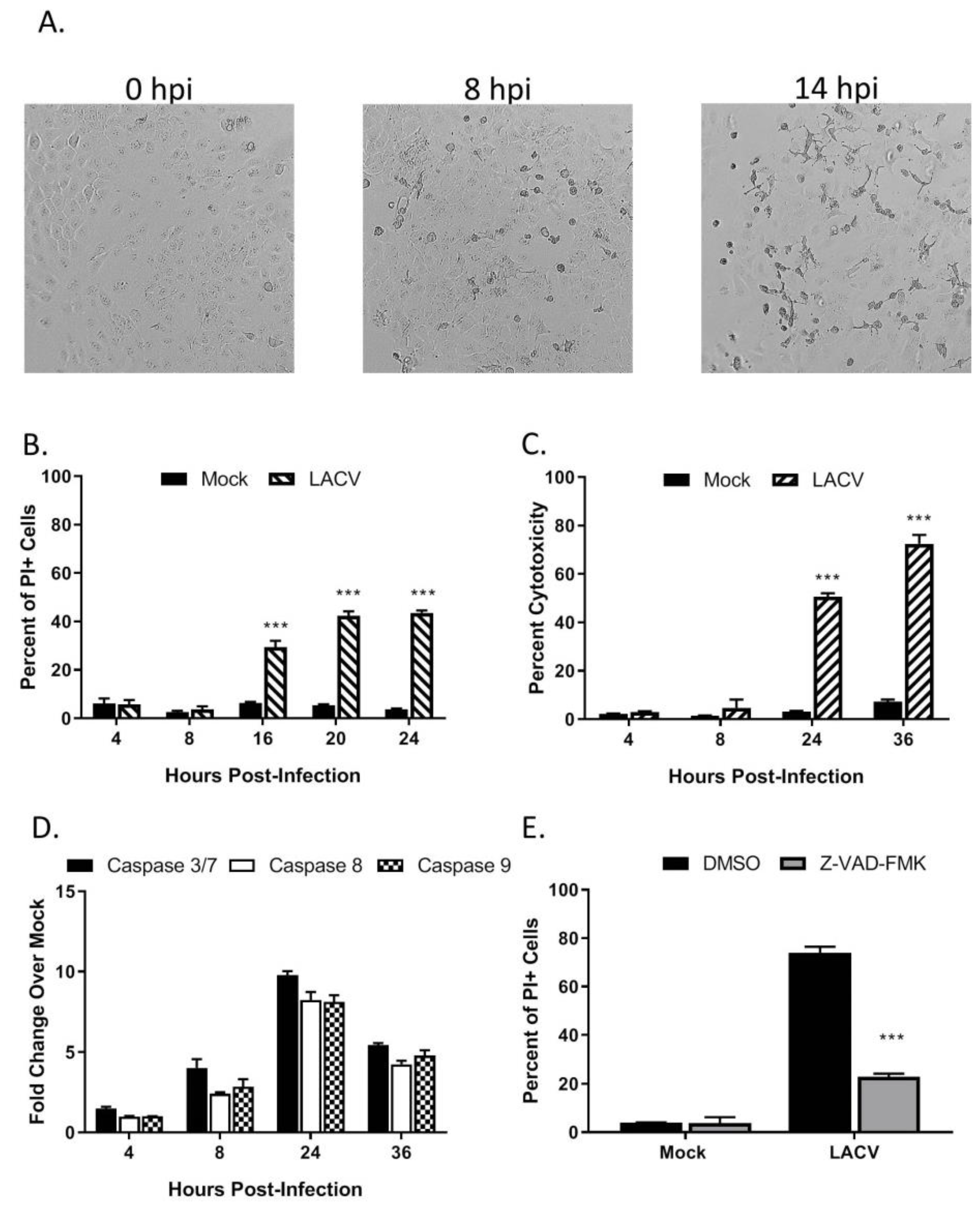
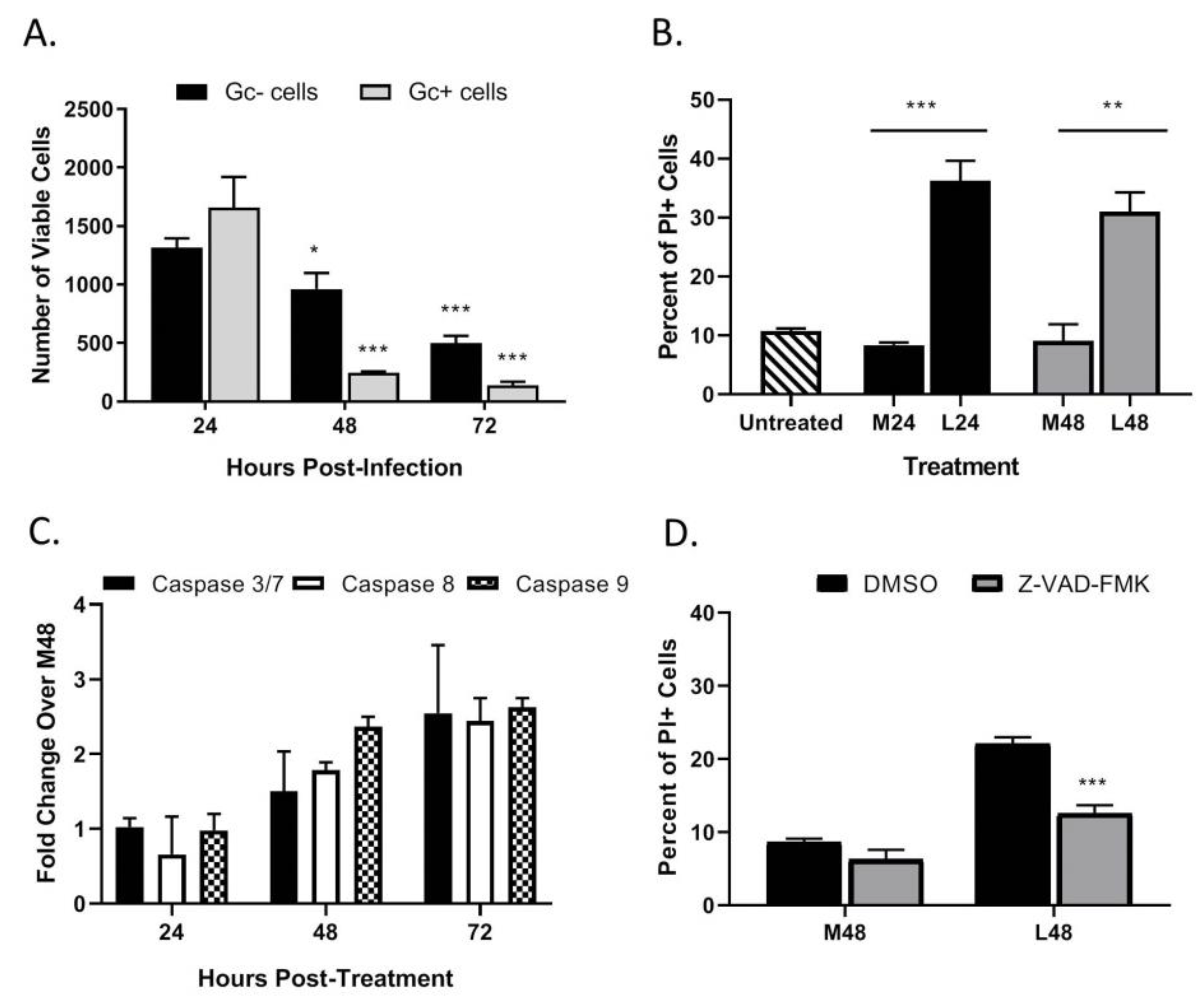
3.2. LACV Infection Induces Caspase-Dependent Cell Death in HaCaT cells
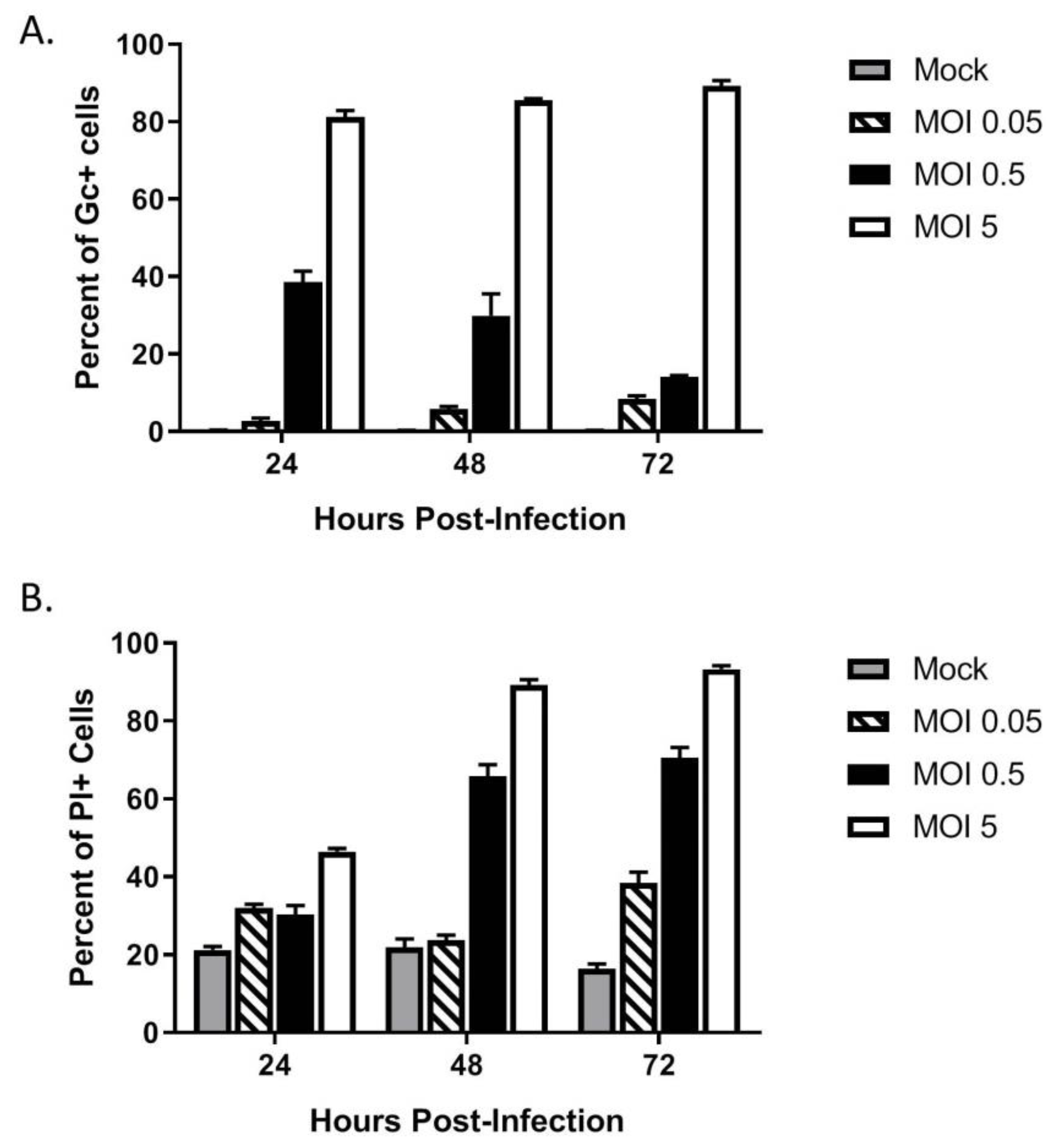
3.3. Multi-Cycle Spread of LACV Infection Is Restricted in HaCaT Cells
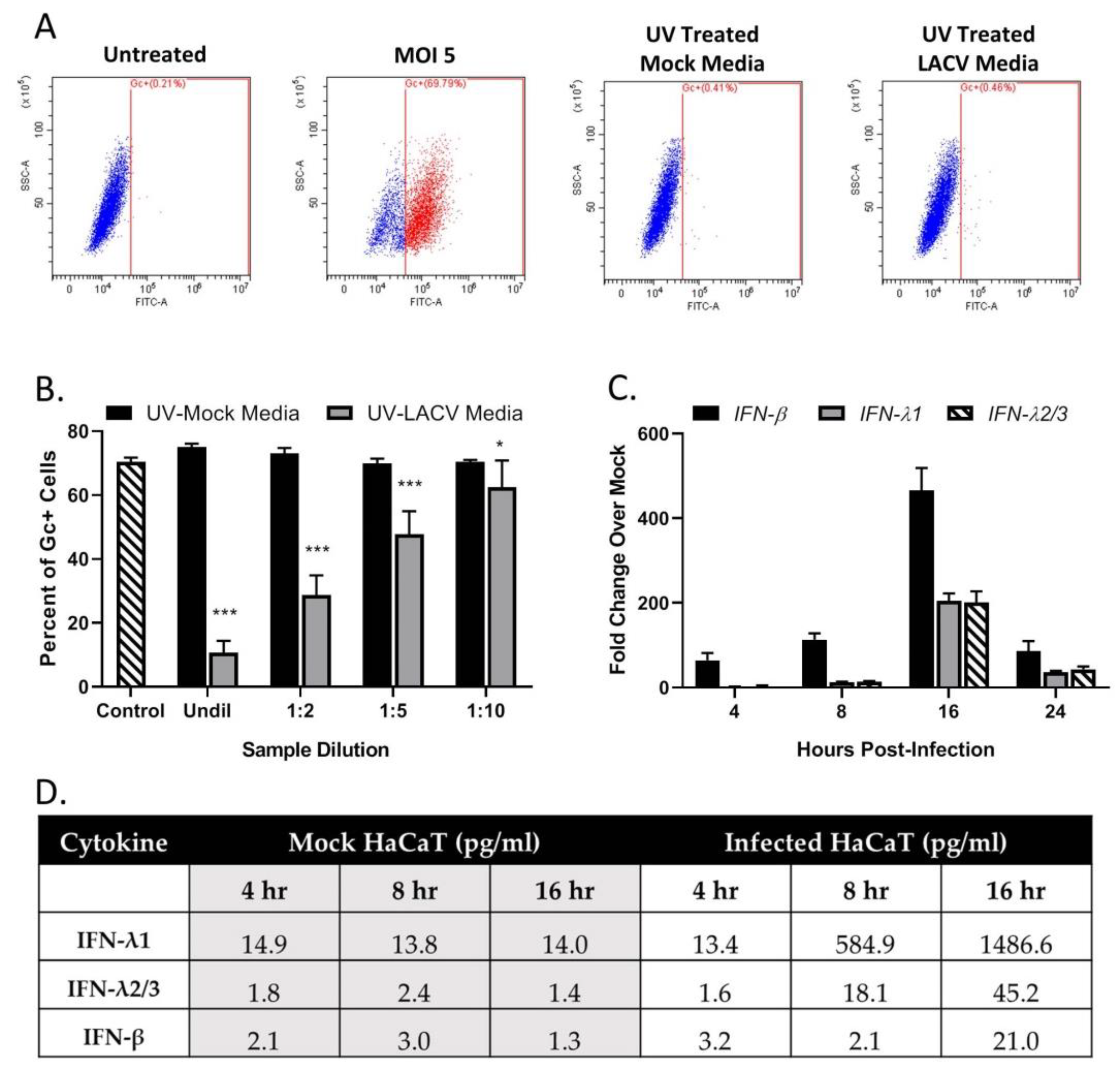
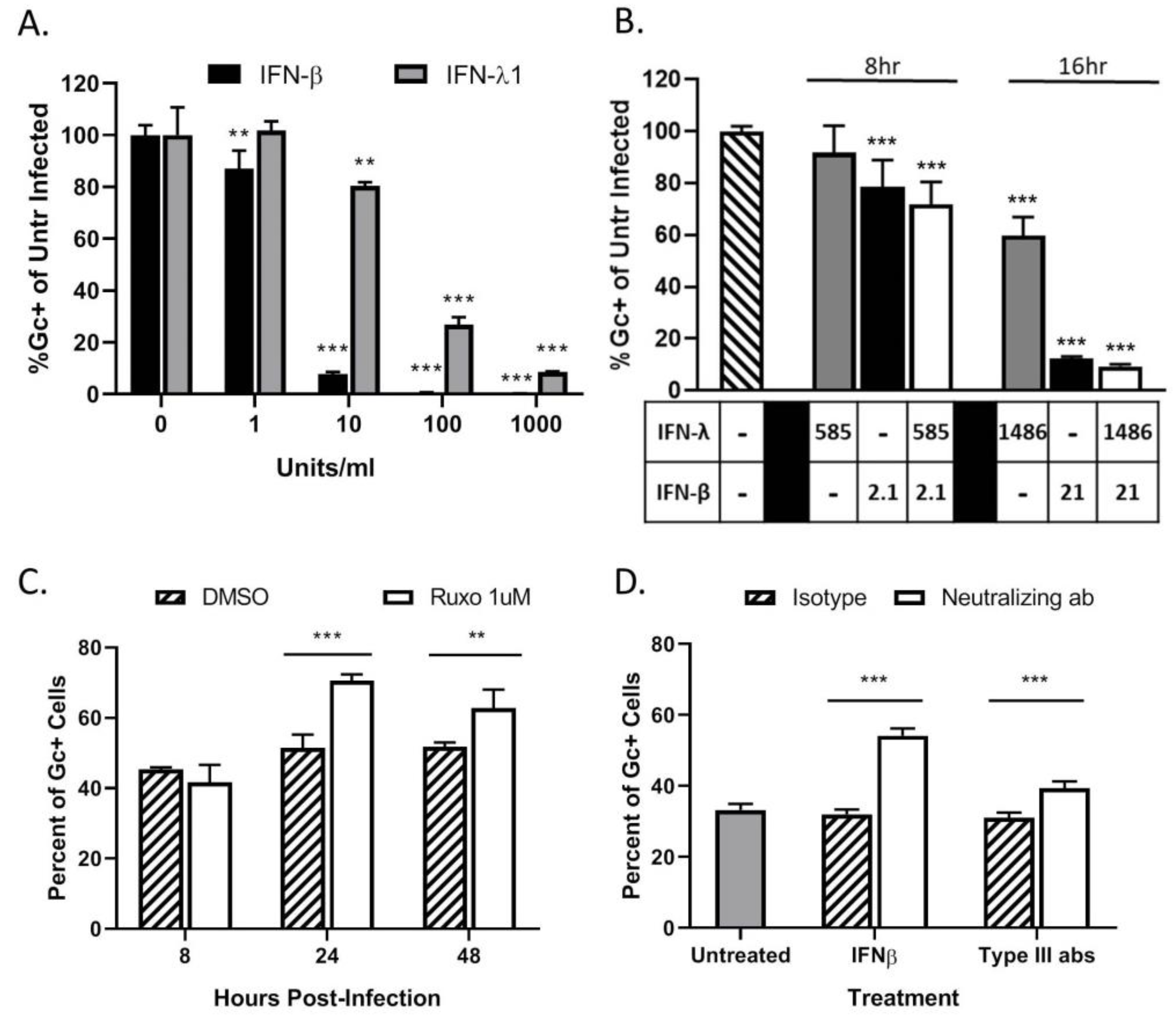
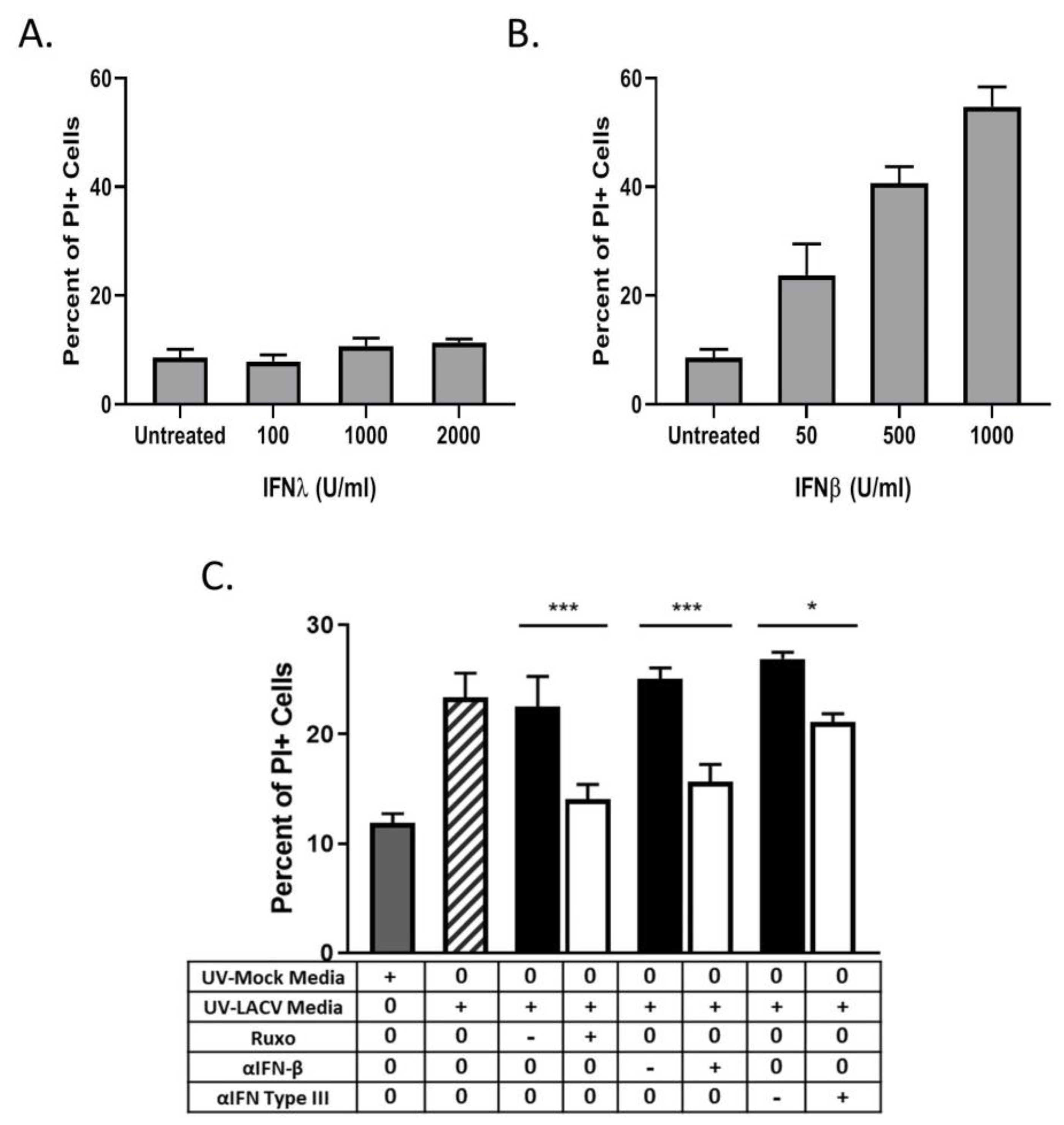
3.4. Restriction in Spread through a Population of HaCaT Cells is Due to an Antiviral Response Primarily Driven by Type I IFN
3.5. LACV Infection Induces Cell Death in Non-Infected Bystander HaCaT Cells
3.6. Bystander Cell Death Observed in Non-Infected HaCaT Cells is IFN-Dependent
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frischknecht, F. The skin as interface in the transmission of arthropod-borne pathogens. Cell. Microbiol. 2007, 9, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M. Molecular biology of the Bunyaviridae. J. Gen. Virol. 1990, 71, 501–522. [Google Scholar] [CrossRef]
- McJunkin, J.E.; Reyes, E.C.d.l.; Irazuzta, J.E.; Caceres, M.J.; Khan, R.R.; Minnich, L.L.; Fu, K.D.; Lovett, G.D.; Tsai, T.; Thompson, A. La Crosse encephalitis in children. N. Engl. J. Med. 2001, 344, 801. [Google Scholar] [PubMed]
- Rust, R.S.; Thompson, W.H.; Matthews, C.G.; Beaty, B.J.; Chun, R.W. La Crosse and other forms of California encephalitis. J. Child Neurol. 1999, 14, 1–14. [Google Scholar] [PubMed]
- ArboNET, Arboviral Diseases Branch, Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/lac/tech/epi.html (accessed on 13 January 2020).
- Evans, A.B.; Peterson, K.E. Throw out the Map: Neuropathogenesis of the Globally Expanding California Serogroup of Orthobunyaviruses. Viruses 2019, 11, 794. [Google Scholar] [CrossRef]
- Grimstad, P.R.; Barrett, C.L.; Humphrey, R.L.; Sinsko, M.J. Serologic evidence for widespread infection with La Crosse and St. Louis encephalitis viruses in the Indiana human population. Am. J. Epidemiol. 1984, 119, 913–930. [Google Scholar] [CrossRef]
- Haddow, A.D.; Odoi, A. The incidence risk, clustering, and clinical presentation of La Crosse virus infections in the eastern United States, 2003–2007. PLoS ONE 2009, 4, e6145. [Google Scholar] [CrossRef]
- Gerhardt, R.R.; Gottfried, K.L.; Apperson, C.S.; Davis, B.S.; Erwin, P.C.; Smith, A.B.; Panella, N.A.; Powell, E.E.; Nasci, R.S. First Isolation of La Crosse Virus from Naturally Infected Aedes albopictus. Emerg. Infect. Dis. 2001, 7, 807. [Google Scholar] [CrossRef]
- Harris, M.C.; Dotseth, E.J.; Jackson, B.T.; Zin, S.D.; Marek, P.E.; Kramer, L.D.; Paulson, S.L.; Hawley, D.M. La Crosse Virus in Aedes Japonicus Mosquitoes in the Appalachian Region, United States. Emerg. Infect. Dis. 2015, 21, 646. [Google Scholar] [CrossRef]
- Taylor, K.G.; Woods, T.A.; Winkler, C.W.; Carmody, A.B.; Peterson, K.E. Age-dependent myeloid dendritic cell responses mediate resistance to la crosse virus-induced neurological disease. J. Virol. 2014, 88, 11070–11079. [Google Scholar] [CrossRef]
- Johnson, R.T. Pathogenesis of La Crosse virus in mice. Prog. Clin. Biol Res. 1983, 123, 139–144. [Google Scholar]
- Griot, C.; Gonzalez-Scarano, F.; Nathanson, N. Molecular determinants of the virulence and infectivity of California serogroup bunyaviruses. Annu. Rev. Microbiol 1993, 47, 117–138. [Google Scholar] [CrossRef]
- Bennett, R.S.; Cress, C.M.; Ward, J.M.; Firestone, C.-Y.; Murphy, B.R.; Whitehead, S.S. La Crosse virus infectivity, pathogenesis, and immunogenicity in mice and monkeys. Virol. J. 2008, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 2014, 5, e01476. [Google Scholar] [CrossRef] [PubMed]
- Proenca-Modena, J.L.; Hyde, J.L.; Sesti-Costa, R.; Lucas, T.; Pinto, A.K.; Richner, J.M.; Gorman, M.J.; Lazear, H.M.; Diamond, M.S. Interferon-Regulatory Factor 5-Dependent Signaling Restricts Orthobunyavirus Dissemination to the Central Nervous System. J. Virol. 2016, 90, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Carlton-Smith, C.; Elliott, R.M. Viperin, MTAP44, and protein kinase R contribute to the interferon-induced inhibition of Bunyamwera Orthobunyavirus replication. J. Virol. 2012, 86, 11548–11557. [Google Scholar] [CrossRef]
- Frese, M.; Kochs, G.; Feldmann, H.; Hertkorn, C.; Haller, O. Inhibition of bunyaviruses, phleboviruses, and hantaviruses by human MxA protein. J. Virol. 1996, 70, 915–923. [Google Scholar]
- Hefti, H.P.; Frese, M.; Landis, H.; Di, P.C.; Aguzzi, A.; Haller, O.; Pavlovic, J. Human MxA protein protects mice lacking a functional alpha/beta interferon system against La crosse virus and other lethal viral infections. J. Virol. 1999, 73, 6984–6991. [Google Scholar]
- Georg, K.; Christian, J.; Heinz, H.; Otto, H. Antivirally Active MxA Protein Sequesters La Crosse Virus Nucleocapsid Protein into Perinuclear Complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 3153. [Google Scholar]
- Lim, P.Y.; Behr, M.J.; Chadwick, C.M.; Shi, P.Y.; Bernard, K.A. Keratinocytes are cell targets of West Nile virus in vivo. J. Virol. 2011, 85, 5197–5201. [Google Scholar] [CrossRef]
- Bernard, E.; Hamel, R.; Neyret, A.; Ekchariyawat, P.; Moles, J.P.; Simmons, G.; Chazal, N.; Despres, P.; Misse, D.; Briant, L. Human keratinocytes restrict chikungunya virus replication at a post-fusion step. Virology 2015, 476, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wichit, S.; Diop, F.; Hamel, R.; Talignani, L.; Ferraris, P.; Cornelie, S.; Liegeois, F.; Thomas, F.; Yssel, H.; Misse, D. Aedes Aegypti saliva enhances chikungunya virus replication in human skin fibroblasts via inhibition of the type I interferon signaling pathway. Infect. Genet. Evol. 2017, 55, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Ekchariyawat, P.; Hamel, R.; Bernard, E.; Wichit, S.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; Choumet, V.; Yssel, H.; Despres, P.; et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 2015, 32, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Puiprom, O.; Vargas, R.E.M.; Potiwat, R.; Chaichana, P.; Ikuta, K.; Ramasoota, P.; Okabayashi, T. Characterization of chikungunya virus infection of a human keratinocyte cell line: Role of mosquito salivary gland protein in suppressing the host immune response. Infect. Genet. Evol. 2013, 17, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Surasombatpattana, P.; Hamel, R.; Patramool, S.; Luplertlop, N.; Thomas, F.; Desprès, P.; Briant, L.; Yssel, H.; Missé, D. Dengue virus replication in infected human keratinocytes leads to activation of antiviral innate immune responses. Infect. Genet. Evol. 2011, 11, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, M.; Meza-Sanchez, D.; Garcia-Cordero, J.; Bustos-Arriaga, J.; Velez-Del Valle, C.; Marsch-Moreno, M.; Castro-Jimenez, T.; Flores-Romo, L.; Santos-Argumedo, L.; Gutierrez-Castaneda, B.; et al. Human keratinocyte cultures (HaCaT) can be infected by DENV, triggering innate immune responses that include IFN-λ and LL37. Immunobiology 2018, 223, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Duangkhae, P.; Erdos, G.; Ryman, K.D.; Watkins, S.C.; Falo Jr, L.D.; Marques Jr, E.T.; Barratt-Boyes, S.M. Interplay between Keratinocytes and Myeloid Cells Drives Dengue Virus Spread in Human Skin. J. Investig. Dermatol. 2018, 138, 618–626. [Google Scholar] [CrossRef]
- Garcia, M.; Wehbe, M.; Leveque, N.; Bodet, C. Skin innate immune response to flaviviral infection. Eur. Cytokine Netw. 2017, 28, 41–51. [Google Scholar] [CrossRef]
- Pivarcsi, A.; Bodai, L.; Réthi, B.; Kenderessy-Szabó, A.; Koreck, A.; Széll, M.; Beer, Z.; Bata-Csörgoo, Z.; Magócsi, M.; Rajnavölgyi, E.; et al. Expression and function of Toll-like receptors 2 and 4 in human keratinocytes. Int. Immunol. 2003, 15, 721–730. [Google Scholar]
- Lebre, M.C.; van der Aar, A.M.; van Baarsen, L.; van Capel, T.M.; Schuitemaker, J.H.; Kapsenberg, M.L.; de Jong, E.C. Human Keratinocytes Express Functional Toll-Like Receptor 3, 4, 5, and 9. J. Investig. Dermatol. 2007, 127, 331–341. [Google Scholar] [CrossRef]
- Kalali, B.N.; Kollisch, G.; Mages, J.; Muller, T.; Bauer, S.; Wagner, H.; Ring, J.; Lang, R.; Mempel, M.; Ollert, M. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J. Immunol. 2008, 181, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Kupper, T.S.; Ballard, D.W.; Chua, A.O.; McGuire, J.S.; Flood, P.M.; Horowitz, M.C.; Langdon, R.; Lightfoot, L.; Gubler, U. Human keratinocytes contain mRNA indistinguishable from monocyte interleukin 1 alpha and beta mRNA. Keratinocyte epidermal cell-derived thymocyte-activating factor is identical to interleukin 1. J. Exp. Med. 1986, 164, 2095–2100. [Google Scholar] [PubMed]
- Larsen, C.G.; Anderson, A.O.; Oppenheim, J.J.; Matsushima, K. Production of interleukin-8 by human dermal fibroblasts and keratinocytes in response to interleukin-1 or tumour necrosis factor. Immunology 1989, 68, 31–36. [Google Scholar] [PubMed]
- Kock, A.; Schwarz, T.; Kirnbauer, R.; Urbanski, A.; Perry, P.; Ansel, J.C.; Luger, T.A. Human keratinocytes are a source for tumor necrosis factor alpha: Evidence for synthesis and release upon stimulation with endotoxin or ultraviolet light. J. Exp. Med. 1990, 172, 1609–1614. [Google Scholar] [CrossRef]
- Oxholm, A.; Diamant, M.; Oxholm, P.; Bendtzen, K. Interleukin-6 and tumour necrosis factor alpha are expressed by keratinocytes but not by Langerhans cells. Apmis 1991, 99, 58–64. [Google Scholar] [CrossRef]
- Grewe, M.; Gyufko, K.; Krutmann, J. Interleukin-10 production by cultured human keratinocytes: Regulation by ultraviolet B and ultraviolet A1 radiation. J. Investig. Dermatol. 1995, 104, 3–6. [Google Scholar]
- Howie, S.E.M.; Aldridge, R.D.; McVittie, E.; Forsey, R.J.; Sands, C.; Hunter, J.A.A. Epidermal keratinocyte production of interferon-gamma immunoreactive protein and mRNA is an early event in allergic contact dermatitis. J. Investig. Dermatol. 1996, 106, 1218–1223. [Google Scholar]
- Fujisawa, H.; Kondo, S.; Wang, B.; Shivji, G.M.; Sauder, D.N. The expression and modulation of IFN-α and IFN-β in human keratinocytes. J. Interferon Cytokine Res. 1997, 17, 721–725. [Google Scholar] [CrossRef]
- Zampetti, A.; Mastrofrancesco, A.; Flori, E.; Maresca, V.; Picardo, M.; Amerio, P.; Feliciani, C. Proinflammatory cytokine production in HaCaT cells treated by eosin: Implications for the topical treatment of psoriasis. Int. J. Immunopathol. Pharm. 2009, 22, 1067–1075. [Google Scholar] [CrossRef]
- Kimura, K.; Matsuzaki, Y.; Nishikawa, Y.; Kitamura, H.; Akasaka, E.; Rokunohe, D.; Nakano, H.; Imaizumi, T.; Satoh, K.; Sawamura, D. Characterization of retinoic acid-inducible gene-I (RIG-I) expression corresponding to viral infection and UVB in human keratinocytes. J. Dermatol. Sci. 2012, 66, 64–70. [Google Scholar] [CrossRef]
- Gil, T.-Y.; Kang, Y.-M.; Eom, Y.-J.; Hong, C.-H.; An, H.-J. Anti-Atopic Dermatitis Effect of Seaweed Fulvescens Extract via Inhibiting the STAT1 Pathway. Mediat. Inflamm. 2019, 1–11. [Google Scholar] [CrossRef]
- Cinque, B.; Di Marzio, L.; Della Riccia, D.N.; Bizzini, F.; Giuliani, M.; Fanini, D.; De Simone, C.; Cifone, M.G. Effect of Bifidobacterium infantis on Interferon- gamma- induced keratinocyte apoptosis: A potential therapeutic approach to skin immune abnormalities. Int. J. Immunopathol. Pharm. 2006, 19, 775–786. [Google Scholar] [CrossRef]
- Deyrieux, A.F.; Wilson, V.G. In vitro culture conditions to study keratinocyte differentiation using the HaCaT cell line. Cytotechnology 2007, 54, 77. [Google Scholar] [CrossRef] [PubMed]
- Colombo, I.; Sangiovanni, E.; Maggio, R.; Mattozzi, C.; Zava, S.; Corbett, Y.; Fumagalli, M.; Carlino, C.; Corsetto, P.A.; Scaccabarozzi, D.; et al. HaCaT Cells as a Reliable In Vitro Differentiation Model to Dissect the Inflammatory/Repair Response of Human Keratinocytes. Mediat. Inflamm. 2017, 1–12. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; García-Sastre, A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001, 12, 143–156. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69. [Google Scholar] [CrossRef]
- Verbruggen, P.; Ruf, M.; Blakqori, G.; Overby, A.K.; Heidemann, M.; Eick, D.; Weber, F. Interferon antagonist NSs of La Crosse virus triggers a DNA damage response-like degradation of transcribing RNA polymerase II. J. Biol. Chem. 2011, 286, 3681–3692. [Google Scholar] [CrossRef]
- Kallfass, C.; Ackerman, A.; Lienenklaus, S.; Weiss, S.; Heimrich, B.; Staeheli, P. Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice. J. Virol. 2012, 86, 11223–11230. [Google Scholar] [CrossRef]
- Meager, A.; Visvalingam, K.; Dilger, P.; Bryan, D.; Wadhwa, M. Biological activity of interleukins-28 and -29: Comparison with type I interferons. Cytokine 2005, 31, 109–118. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Mukherjee, P.; Woods, T.A.; Moore, R.A.; Peterson, K.E. Activation of the Innate Signaling Molecule MAVS by Bunyavirus Infection Upregulates the Adaptor Protein SARM1, Leading to Neuronal Death. Immunity 2013, 38, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Colon-Ramos, D.A.; Irusta, P.M.; Gan, E.C.; Olson, M.R.; Song, J.; Morimoto, R.I.; Elliott, R.M.; Lombard, M.; Hollingsworth, R.; Hardwick, J.M.; et al. Inhibition of translation and induction of apoptosis by Bunyaviral nonstructural proteins bearing sequence similarity to Reaper. Mol. Biol. Cell 2003, 14, 4162–4172. [Google Scholar] [CrossRef] [PubMed]
- Castelli, J.C.; Hassel, B.A.; Wood, K.A.; Li, X.-L.; Amemiya, K.; Dalakas, M.C.; Torrence, P.F.; Youle, R.J. A study of the interferon antiviral mechanism: Apoptosis activation by the 2-5A system. J. Exp. Med. 1997, 186, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Dedoni, S.; Olianas, M.C.; Onali, P. Interferon-β induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J. Neurochem. 2010, 115, 1421. [Google Scholar] [CrossRef]
- Apelbaum, A.; Yarden, G.; Warszawski, S.; Harari, D.; Schreiber, G.H. Type I interferons induce apoptosis by balancing cFLIP and caspase-8 independent of death ligands. Mol. Cell Biol 2013, 33, 800–814. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward Primer | Reverse Primer | |
|---|---|---|
| β-actin | 5′-GATCATTGCTCCTCCTGAGC-3′, | 5’-ACTCCTGCTTGCTGATCCAC-3, |
| IFN-β | 5’-CAGCTCTTTCCATGAGCTACAA-3’ | 5’-CAGTATTCAAGCCTCCCATTCA-3’ |
| IFN-λ1 | 5’-CTTGGACCGTGGTGCTG-3’ | 5’-CAGCCCTTCCCAGTTGTG-3’ |
| IFN-λ2/3 | 5’-CAGTGCTGGTGCTGATG-3’ | 5’-GACTTGAACTGGGCTATGTG-3’ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, M.A.; Parks, G.D. La Crosse Virus Infection of Human Keratinocytes Leads to Interferon-Dependent Apoptosis of Bystander Non-Infected Cells In Vitro. Viruses 2020, 12, 253. https://doi.org/10.3390/v12030253
Cruz MA, Parks GD. La Crosse Virus Infection of Human Keratinocytes Leads to Interferon-Dependent Apoptosis of Bystander Non-Infected Cells In Vitro. Viruses. 2020; 12(3):253. https://doi.org/10.3390/v12030253
Chicago/Turabian StyleCruz, Maria A., and Griffith D. Parks. 2020. "La Crosse Virus Infection of Human Keratinocytes Leads to Interferon-Dependent Apoptosis of Bystander Non-Infected Cells In Vitro" Viruses 12, no. 3: 253. https://doi.org/10.3390/v12030253
APA StyleCruz, M. A., & Parks, G. D. (2020). La Crosse Virus Infection of Human Keratinocytes Leads to Interferon-Dependent Apoptosis of Bystander Non-Infected Cells In Vitro. Viruses, 12(3), 253. https://doi.org/10.3390/v12030253