Characteristics of Human OAS1 Isoform Proteins



Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Cloning and Expression of hOAS1 Isoform Proteins in Bacteria
2.3. In Vitro 2′-5′ OAS Activity Assay
2.4. Functional Analysis of hOAS1 Isoforms in Mammalian Cells
2.5. Yeast Two Hybrid Assay
2.6. Yeast Co-Transformation
2.7. In Vitro Transcription/Translation and Pull-Down Assay
2.8. Mammalian Cell Co-Immunoprecipitation
2.9. Western Blot Assay
3. Results
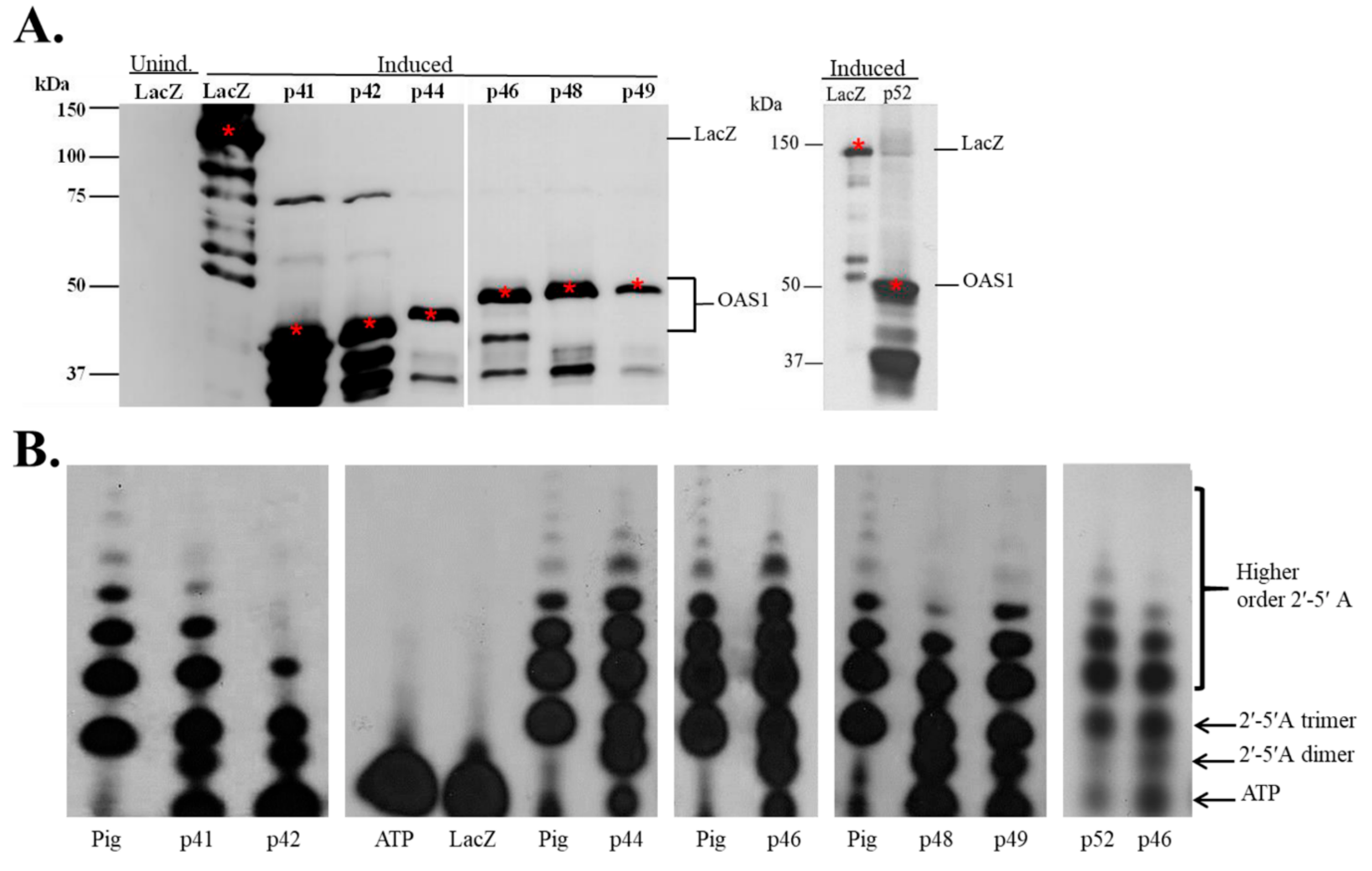
3.1. In Vitro Expression of Recombinant hOAS1 Isoforms
3.2. Recombinant hOAS1 p41, p42, p44, p46, p48, p49 and p52 Isoforms Are Functionally Active
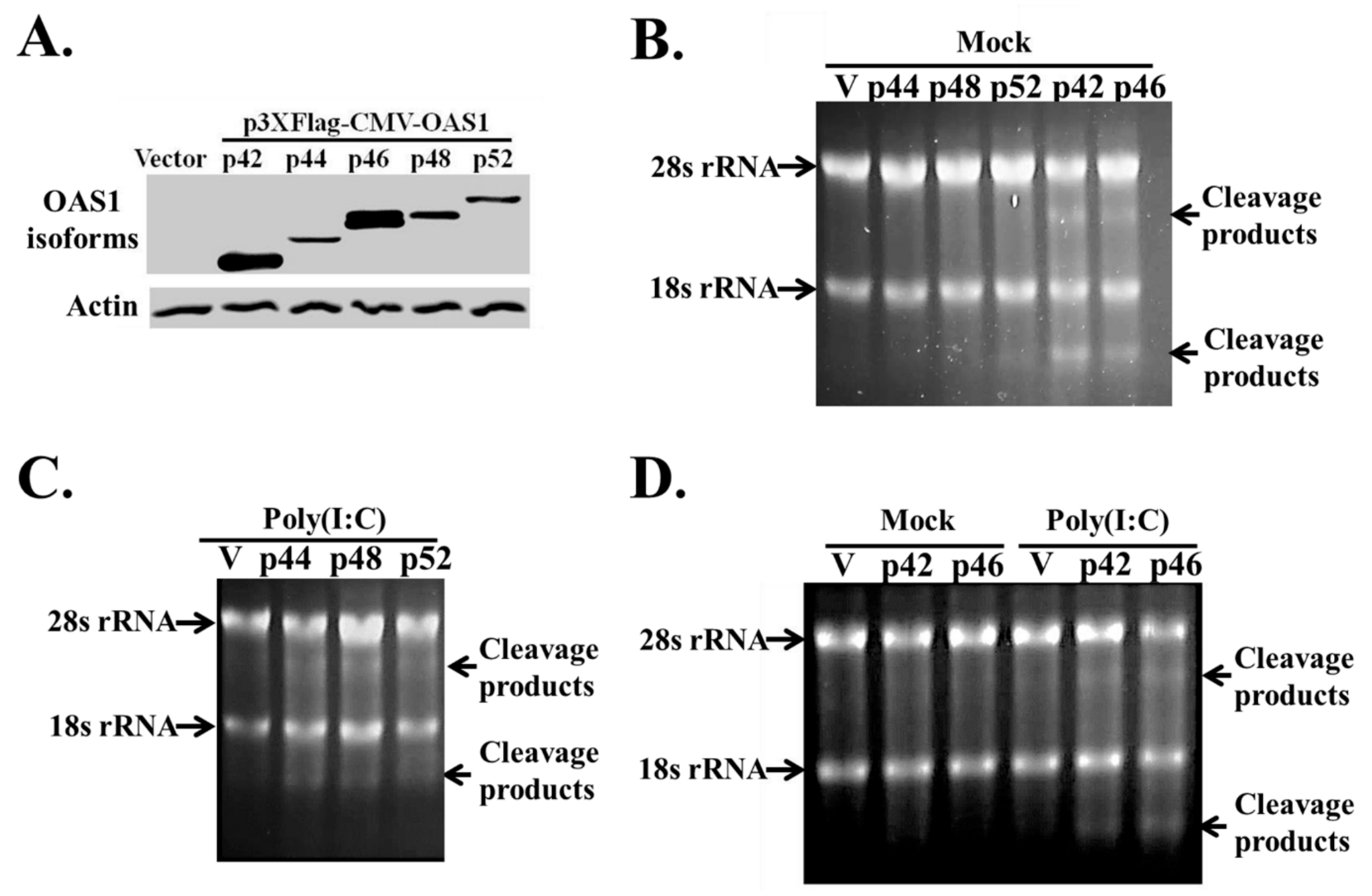
3.3. The hOAS1 p42, p44, p46, p48 and p52 Isoforms Are Functionally Active in Mammalian Cells
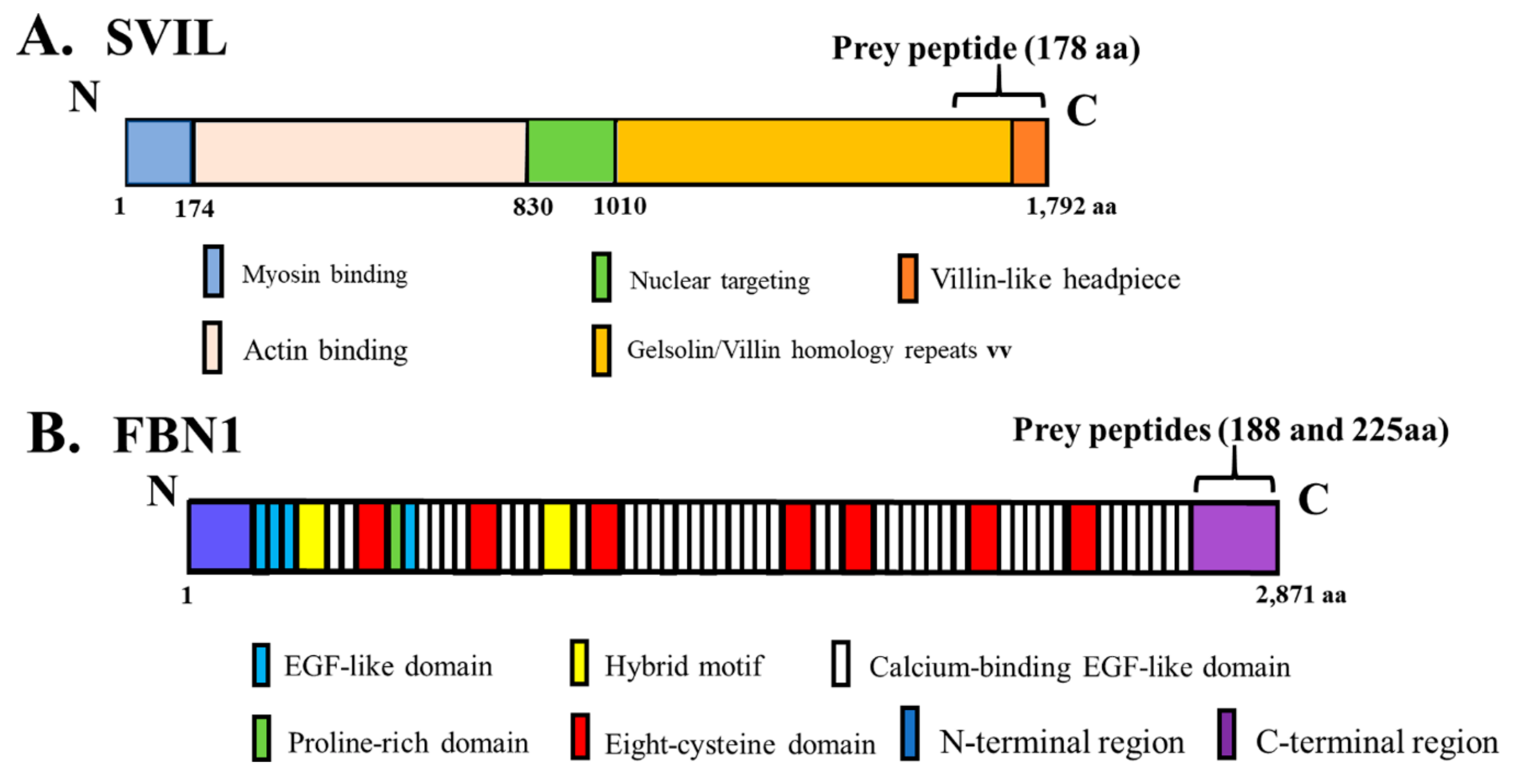
3.4. Identification of Novel Binding Partners for hOAS1 p42 and p44 Isoforms in a Yeast Two-Hybrid Assay
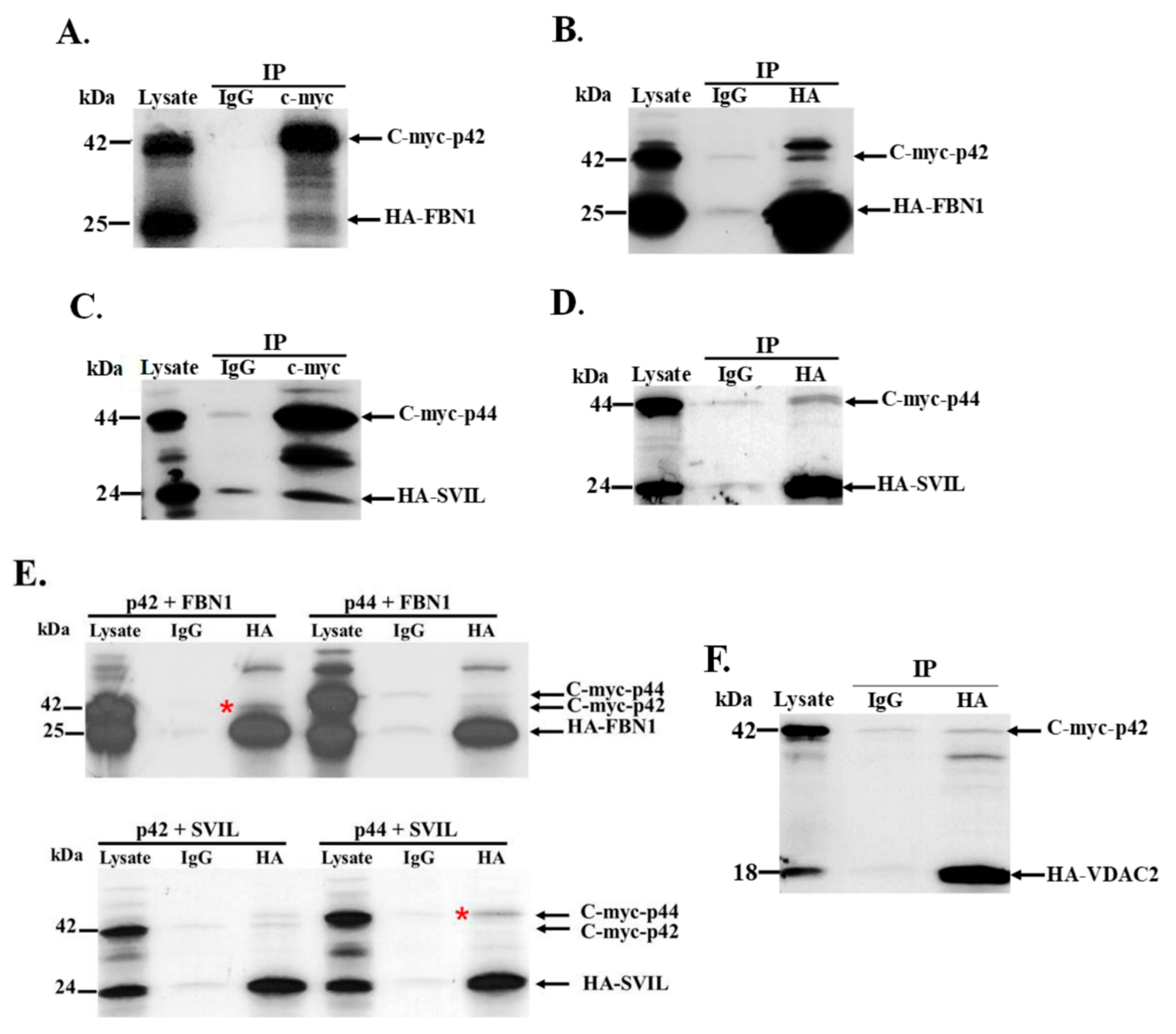
3.5. Interaction of In Vitro Synthesized hOAS1 p42 and p44 Isoform Proteins and Their Identified Binding Partners
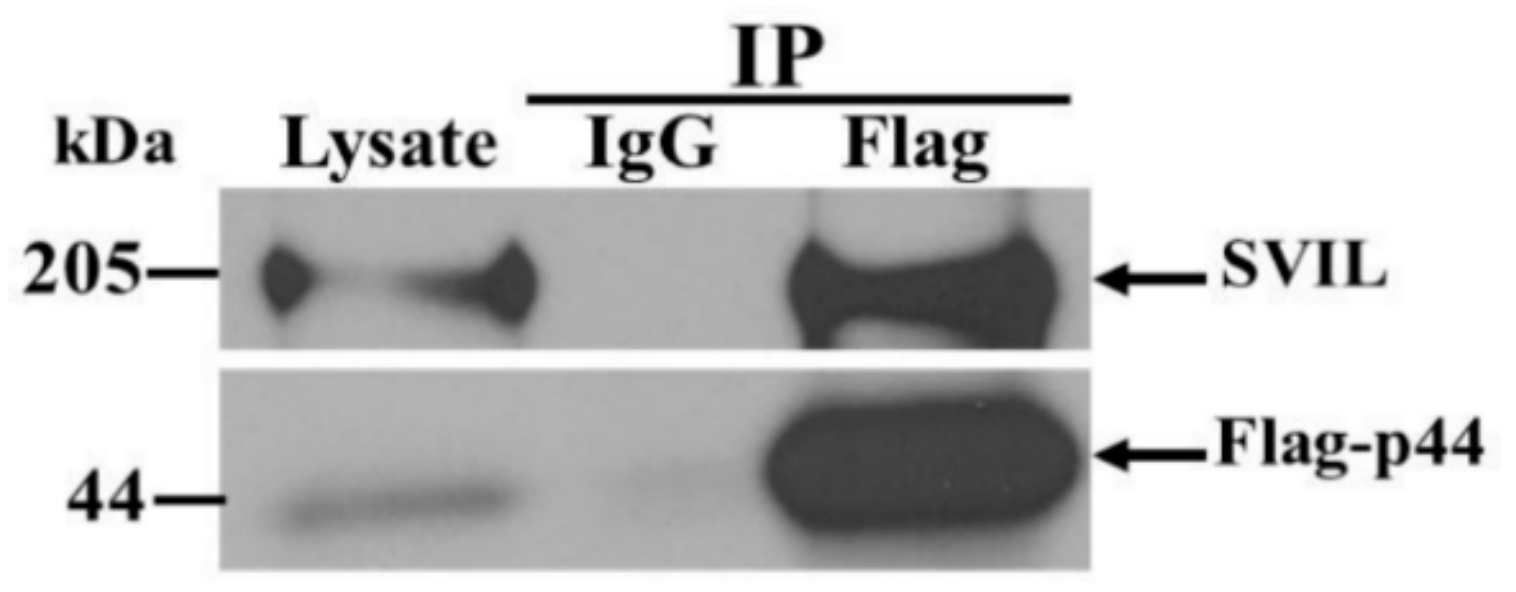
3.6. The hOAS1 p44 Isoform Interacts with Endogenous SVIL Protein in Mammalian Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kristiansen, H.; Gad, H.H.; Eskildsen-Larsen, S.; Despres, P.; Hartmann, R. The oligoadenylate synthetase family: An ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2011, 31, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, K.H.; Poulsen, J.B.; Reintamm, T.; Saby, E.; Martensen, P.M.; Kelve, M.; Justesen, J. Evolution of the 2’–5’-oligoadenylate synthetase family in eukaryotes and bacteria. J. Mol. Evol. 2009, 69, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Rebouillat, D.; Marie, I.; Hovanessian, A.G. Molecular cloning and characterization of two related and interferon-induced 56-kDa and 30-kDa proteins highly similar to 2′–5′ oligoadenylate synthetase. Eur. J. Biochem. 1998, 257, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Hovanessian, A.G. The 69-kDa 2-5A synthetase is composed of two homologous and adjacent functional domains. J. Biol. Chem. 1992, 267, 9933–9939. [Google Scholar] [PubMed]
- Hartmann, R.; Olsen, H.S.; Widder, S.; Jorgensen, R.; Justesen, J. p59OASL, a 2′–5′ oligoadenylate synthetase like protein: A novel human gene related to the 2′–5′ oligoadenylate synthetase family. Nucleic Acids Res. 1998, 26, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Justesen, J. The human 2′–5′oligoadenylate synthetase family: Unique interferon-inducible enzymes catalyzing 2’–5’ instead of 3′–5′ phosphodiester bond formation. Biochimie 2007, 89, 779–788. [Google Scholar] [CrossRef]
- Dong, B.; Xu, L.; Zhou, A.; Hassel, B.A.; Lee, X.; Torrence, P.F.; Silverman, R.H. Intrinsic molecular activities of the interferon-induced 2-5A-dependent RNase. J. Biol. Chem. 1994, 269, 14153–14158. [Google Scholar]
- Player, M.R.; Torrence, P.F. The 2–5A system: Modulation of viral and cellular processes through acceleration of RNA degradation. Pharmacol. Ther. 1998, 78, 55–113. [Google Scholar] [CrossRef]
- Ibsen, M.S.; Gad, H.H.; Andersen, L.L.; Hornung, V.; Julkunen, I.; Sarkar, S.N.; Hartmann, R. Structural and functional analysis reveals that human OASL binds dsRNA to enhance RIG-I signaling. Nucleic Acids Res. 2015, 43, 5236–5248. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef]
- Justesen, J.; Hartmann, R.; Kjeldgaard, N.O. Gene structure and function of the 2′–5′-oligoadenylate synthetase family. Cell. Mol. Life Sci. CMLS 2000, 57, 1593–1612. [Google Scholar] [CrossRef] [PubMed]
- Bonnevie-Nielsen, V.; Field, L.L.; Lu, S.; Zheng, D.J.; Li, M.; Martensen, P.M.; Nielsen, T.B.; Beck-Nielsen, H.; Lau, Y.L.; Pociot, F. Variation in antiviral 2′,5′-oligoadenylate synthetase (2′5′AS) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the OAS1 gene. Am. J. Hum. Genet. 2005, 76, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.E.; Gewert, D.R.; Tugwell, M.E.; McMahon, M.; Williams, B.R. Human 2–5A synthetase: Characterization of a novel cDNA and corresponding gene structure. EMBO J. 1985, 4, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Kusari, J.; Bandyopadhyay, S.K.; Samanta, H.; Kumar, R.; Sen, G.C. Cloning, sequencing, and expression of two murine 2’–5’-oligoadenylate synthetases. Structure-function relationships. J. Biol. Chem. 1991, 266, 15293–15299. [Google Scholar]
- Benech, P.; Mory, Y.; Revel, M.; Chebath, J. Structure of two forms of the interferon-induced (2′–5′) oligo A synthetase of human cells based on cDNAs and gene sequences. EMBO J. 1985, 4, 2249–2256. [Google Scholar] [CrossRef]
- Ghosh, A.; Sarkar, S.N.; Rowe, T.M.; Sen, G.C. A specific isozyme of 2′–5′ oligoadenylate synthetase is a dual function proapoptotic protein of the Bcl-2 family. J. Biol. Chem. 2001, 276, 25447–25455. [Google Scholar] [CrossRef]
- Deo, S.; Patel, T.R.; Dzananovic, E.; Booy, E.P.; Zeid, K.; McEleney, K.; Harding, S.E.; McKenna, S.A. Activation of 2′ 5′-oligoadenylate synthetase by stem loops at the 5′-end of the West Nile virus genome. PLoS ONE 2014, 9, e92545. [Google Scholar] [CrossRef]
- Lin, R.J.; Yu, H.P.; Chang, B.L.; Tang, W.C.; Liao, C.L.; Lin, Y.L. Distinct antiviral roles for human 2′,5′-oligoadenylate synthetase family members against dengue virus infection. J. Immunol. 2009, 183, 8035–8043. [Google Scholar] [CrossRef]
- Elbahesh, H.; Jha, B.K.; Silverman, R.H.; Scherbik, S.V.; Brinton, M.A. The Flvr-encoded murine oligoadenylate synthetase 1b (Oas1b) suppresses 2-5A synthesis in intact cells. Virology 2011, 409, 262–270. [Google Scholar] [CrossRef]
- Hartmann, R.; Justesen, J.; Sarkar, S.N.; Sen, G.C.; Yee, V.C. Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein 2′–5′-oligoadenylate synthetase. Mol. Cell 2003, 12, 1173–1185. [Google Scholar] [CrossRef]
- Hartmann, R.; Norby, P.L.; Martensen, P.M.; Jorgensen, P.; James, M.C.; Jacobsen, C.; Moestrup, S.K.; Clemens, M.J.; Justesen, J. Activation of 2′–5′ oligoadenylate synthetase by single-stranded and double-stranded RNA aptamers. J. Biol. Chem. 1998, 273, 3236–3246. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, J.B.; Kjaer, K.H.; Justesen, J.; Martensen, P.M. Enzyme assays for synthesis and degradation of 2-5As and other 2′–5′ oligonucleotides. BMC Biochem. 2015, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Carey, C.M.; Govande, A.A.; Cooper, J.M.; Hartley, M.K.; Kranzusch, P.J.; Elde, N.C. Recurrent Loss-of-Function Mutations Reveal Costs to OAS1 Antiviral Activity in Primates. Cell Host Microbe 2019, 25, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Reksten, T.R.; Ice, J.A.; Kelly, J.A.; Adrianto, I.; Rasmussen, A.; Wang, S.; He, B.; Grundahl, K.M.; Glenn, S.B.; et al. Identification of a Sjogren’s syndrome susceptibility locus at OAS1 that influences isoform switching, protein expression, and responsiveness to type I interferons. PLoS Genet. 2017, 13, e1006820. [Google Scholar] [CrossRef] [PubMed]
- Courtney, S.C.; Di, H.; Stockman, B.M.; Liu, H.; Scherbik, S.V.; Brinton, M.A. Identification of novel host cell binding partners of Oas1b, the protein conferring resistance to flavivirus-induced disease in mice. J. Virol. 2012, 86, 7953–7963. [Google Scholar] [CrossRef]
- McAveney, K.M.; Book, M.L.; Ling, P.; Chebath, J.; Yu-Lee, L. Association of 2′,5′-oligoadenylate synthetase with the prolactin (PRL) receptor: Alteration in PRL-inducible stat1 (signal transducer and activator of transcription 1) signaling to the IRF-1 (interferon-regulatory factor 1) promoter. Mol. Endocrinol. 2000, 14, 295–306. [Google Scholar]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef]
- Lohofener, J.; Steinke, N.; Kay-Fedorov, P.; Baruch, P.; Nikulin, A.; Tishchenko, S.; Manstein, D.J.; Fedorov, R. The Activation Mechanism of 2′–5′-Oligoadenylate Synthetase Gives New Insights Into OAS/cGAS Triggers of Innate Immunity. Structure 2015, 23, 851–862. [Google Scholar] [CrossRef]
- Donovan, J.; Dufner, M.; Korennykh, A. Structural basis for cytosolic double-stranded RNA surveillance by human oligoadenylate synthetase 1. Proc. Natl. Acad. Sci. USA 2013, 110, 1652–1657. [Google Scholar] [CrossRef]
- Lim, J.K.; Lisco, A.; McDermott, D.H.; Huynh, L.; Ward, J.M.; Johnson, B.; Johnson, H.; Pape, J.; Foster, G.A.; Krysztof, D.; et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 2009, 5, e1000321. [Google Scholar] [CrossRef]
- Whelan, J.N.; Li, Y.; Silverman, R.H.; Weiss, S.R. Zika Virus Production Is Resistant to RNase L Antiviral Activity. J. Virol. 2019, 93, e00313–e00319. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Sarkar, S.N.; Guo, W.; Bandyopadhyay, S.; Sen, G.C. Enzymatic activity of 2′–5′-oligoadenylate synthetase is impaired by specific mutations that affect oligomerization of the protein. J. Biol. Chem. 1997, 272, 33220–33226. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, K.H.; Pahus, J.; Hansen, M.F.; Poulsen, J.B.; Christensen, E.I.; Justesen, J.; Martensen, P.M. Mitochondrial localization of the OAS1 p46 isoform associated with a common single nucleotide polymorphism. BMC Cell Biol. 2014, 15, 33. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Dias, H.F.; Osuch, I.H.; Lowe, E.D.; Jensen, S.A.; Redfield, C.; Handford, P.A. The N-Terminal Region of Fibrillin-1 Mediates a Bipartite Interaction with LTBP1. Structure 2017, 25, 1208–1221. [Google Scholar] [CrossRef]
- Wipff, J.; Allanore, Y.; Boileau, C. Interactions between fibrillin-1 and tgf-beta: Consequences and human pathology. Med. Sci. M/S 2009, 25, 161–167. [Google Scholar] [CrossRef]
- Hubmacher, D.; Reinhardt, D.P. One more piece in the fibrillin puzzle. Structure 2009, 17, 635–636. [Google Scholar] [CrossRef]
- Kristiansen, H.; Scherer, C.A.; McVean, M.; Iadonato, S.P.; Vends, S.; Thavachelvam, K.; Steffensen, T.B.; Horan, K.A.; Kuri, T.; Weber, F.; et al. Extracellular 2′–5′ oligoadenylate synthetase stimulates RNase L-independent antiviral activity: A novel mechanism of virus-induced innate immunity. J. Virol. 2010, 84, 11898–11904. [Google Scholar] [CrossRef]
- Thavachelvam, K.; Gad, H.H.; Ibsen, M.S.; Despres, P.; Hokland, M.; Hartmann, R.; Kristiansen, H. Rapid Uptake and Inhibition of Viral Propagation by Extracellular OAS1. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2015, 35, 359–366. [Google Scholar] [CrossRef]
- Giannelli, G.; Antonelli, G.; Fera, G.; Dianzani, F.; Schiraldi, O. 2′,5′-Oligoadenylate synthetase activity as a responsive marker during interferon therapy for chronic hepatitis C. J. Interferon Res. 1993, 13, 57–60. [Google Scholar] [CrossRef]
- Ezelle, H.J.; Malathi, K.; Hassel, B.A. The Roles of RNase-L in Antimicrobial Immunity and the Cytoskeleton-Associated Innate Response. Int. J. Mol. Sci. 2016, 17, 74. [Google Scholar] [CrossRef]
- Khurana, S.; George, S.P. Regulation of cell structure and function by actin-binding proteins: villin’s perspective. FEBS Lett. 2008, 582, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Pope, R.K.; Pestonjamasp, K.N.; Smith, K.P.; Wulfkuhle, J.D.; Strassel, C.P.; Lawrence, J.B.; Luna, E.J. Cloning, characterization, and chromosomal localization of human superillin (SVIL). Genomics 1998, 52, 342–351. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hOAS1 Bait | Peptide Candidate Size (aa) | Putative Protein Containing the Peptide |
|---|---|---|
| p42 | 188, 225 | Fibrillin 1 (FBN1) |
| 95 | Phosphoglucomutase 1 (PGM1) | |
| 127 | Voltage-dependent anion channel 2 (VDAC2) | |
| 120 | Trafficking protein particle complex 8 (TRAPPC8) | |
| 115 | Lysyl Oxidase-Like 3 (LOXL3) | |
| p44 | 178 | Supervillin (SVIL) |
| 206 | G elongation factor, mitochondrial 2 | |
| 139 | COX11 cytochrome c oxidase assembly homolog |
| Peptide Candidate | Co-Transformed Bait | DDO a | TDO/A a | QDO/A a |
|---|---|---|---|---|
| FBN1 | p42 | >100 b | 7 b | 1 b |
| Empty vector | >100 | - | - | |
| PGM1 | p42 | >100 | 8 | 1 |
| Empty vector | >100 | 2 | 2 | |
| VDAC2 | p42 | >100 | 17 | - |
| Empty vector | >100 | - | - | |
| KIAA1012 | p42 | >100 | - | - |
| Empty vector | >100 | - | - | |
| LOXL3 | p44 | >100 | - | - |
| Empty vector | >100 | - | - | |
| SVIL | p44 | >100 | 6 | 1 |
| Empty vector | >100 | - | - | |
| G elongation factor | p44 | >100 | - | - |
| Empty vector | >100 | - | - | |
| COX11 | p44 | >100 | - | - |
| Empty vector | >100 | - | - | |
| T antigen | p53 c | >100 | 43 | 14 |
| T antigen | Lambda d | 28 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di, H.; Elbahesh, H.; Brinton, M.A. Characteristics of Human OAS1 Isoform Proteins. Viruses 2020, 12, 152. https://doi.org/10.3390/v12020152
Di H, Elbahesh H, Brinton MA. Characteristics of Human OAS1 Isoform Proteins. Viruses. 2020; 12(2):152. https://doi.org/10.3390/v12020152
Chicago/Turabian StyleDi, Han, Husni Elbahesh, and Margo A. Brinton. 2020. "Characteristics of Human OAS1 Isoform Proteins" Viruses 12, no. 2: 152. https://doi.org/10.3390/v12020152
APA StyleDi, H., Elbahesh, H., & Brinton, M. A. (2020). Characteristics of Human OAS1 Isoform Proteins. Viruses, 12(2), 152. https://doi.org/10.3390/v12020152