Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Illumina Sequencing and Data Processing
2.3. Recovery of Complete Genomes of Genomoviruses and Unclassified CRESS DNA Viruses
2.4. Viral Sequence Analysis
2.5. Phylogenetic Analysis
2.5.1. Genomoviruses
2.5.2. Unclassified CRESS DNA Viruses
2.5.3. Microviruses
2.6. Recombination Analysis of Genomoviruses
3. Results and Discussion
3.1. Identification of ssDNA Virus Genomes in Sonoran Desert Tortoise Fecal Samples
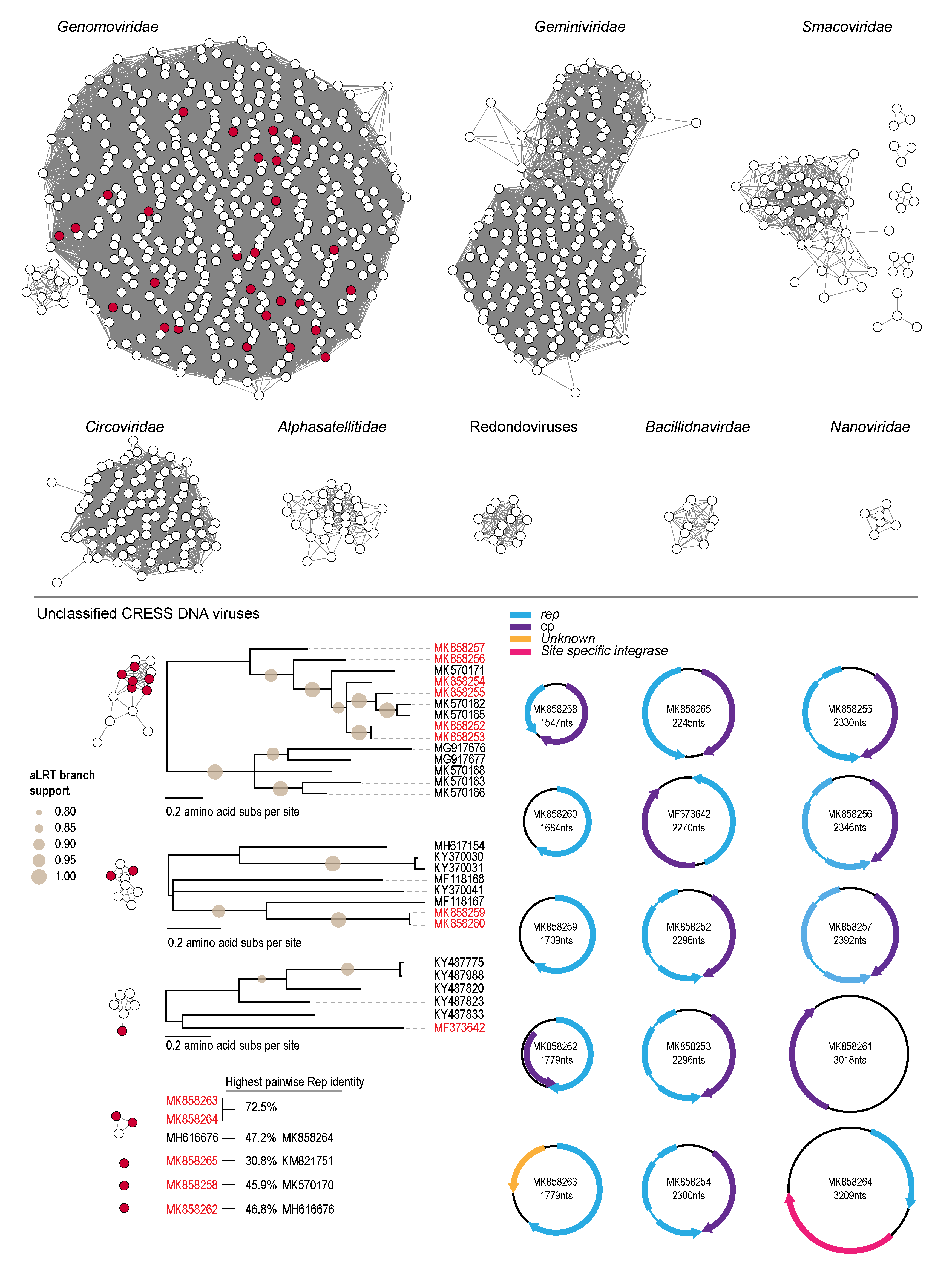
3.2. Genomoviruses
3.3. Unclassified Eukaryotic CRESS DNA Viruses and Circular DNA Molecules
3.4. Microviruses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Medica, P.A.; Nussear, K.E.; Esque, T.C.; Saethre, M.B. Long-Term Growth of Desert Tortoises (Gopherus agassizii) in a Southern Nevada Population. J. Herpetol. 2012, 46, 213–220. [Google Scholar] [CrossRef]
- Curtin, A.J.; Curtin, A.J.; Zug, G.R.; Spotila, J.R. Longevity and growth strategies of the desert tortoise (Gopherus agassizii) in two American deserts. J. Arid Environ. 2009, 73, 463–471. [Google Scholar] [CrossRef]
- Bailey, S.J.; Schwalbe, C.R.; Lowe, C.H. Hibernaculum Use by a Population of Desert Tortoises (Gopherus-Agassizii) in the Sonoran Desert. J. Herpetol. 1995, 29, 361–369. [Google Scholar] [CrossRef]
- Nagy, K.A.; Medica, P.A. Physiological Ecology of Desert Tortoises in Southern Nevada. Herpetologica 1986, 42, 73–92. [Google Scholar]
- Grover, M.C.; DeFalco, L.A. Desert Tortoise (Gopherus Agassizii): Status-of-Knowledge Outline with References; Gen. Tech. Rep. INT-GTR-316; US Department of Agriculture, Intermountain Research Station: Ogden, UT, USA, 1995; Volume 316, p. 134.
- Drake, K.K.; Bowen, L.; Nussear, K.E.; Esque, T.C.; Berger, A.J.; Custer, N.A.; Waters, S.C.; Johnson, J.D.; Miles, A.K.; Lewison, R.L. Negative impacts of invasive plants on conservation of sensitive desert wildlife. Ecosphere 2016, 7, e01531. [Google Scholar] [CrossRef]
- Murphy, R.W.; Berry, K.H.; Edwards, T.; Leviton, A.E.; Lathrop, A.; Riedle, J.D. The dazed and confused identity of Agassiz’s land tortoise, Gopherus agassizii (Testudines, Testudinidae) with the description of a new species, and its consequences for conservation. ZooKeys 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.; Karl, A.E.; Vaughn, M.; Rosen, P.C.; Torres, C.M.; Murphy, R.W. The desert tortoise trichotomy: Mexico hosts a third, new sister-species of tortoise in the Gopherus morafkai–G. agassizii group. ZooKeys 2016, 131. [Google Scholar] [CrossRef]
- Dolby, G.A.; Dorsey, R.J.; Graham, M.R. A legacy of geo-climatic complexity and genetic divergence along the lower Colorado River: Insights from the geological record and 33 desert-adapted animals. J. Biogeogr. 2019, 46, 2479–2505. [Google Scholar] [CrossRef]
- Brown, M.B.; Schumacher, I.M.; Klein, P.A.; Harris, K.; Correll, T.; Jacobson, E.R. Mycoplasma agassizii causes upper respiratory tract disease in the desert tortoise. Infect. Immun. 1994, 62, 4580–4586. [Google Scholar] [CrossRef] [PubMed]
- Service, U.F.a.W. Species Status Assessment for the Sonoran Desert Tortoise; Version 1.0, September 2015; Southwest Region US Fish and Wildlife Service: Albuquerque, NM, USA, 2015.
- Dickinson, V.M.; Schumacher, I.M.; Jarchow, J.L.; Duck, T.; Schwalbe, C.R. Mycoplasmosis in free-ranging desert tortoises in Utah and Arizona. J. Wildl. Dis. 2005, 41, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.A. Mycoplasma agassizii in the Sonoran population of the desert tortoise in Arizona. Master’s Thesis, The University of Arizona, Tucson, AZ, USA, 2008. [Google Scholar]
- Ng, T.F.; Wellehan, J.F.; Coleman, J.K.; Kondov, N.O.; Deng, X.; Waltzek, T.B.; Reuter, G.; Knowles, N.J.; Delwart, E. A tortoise-infecting picornavirus expands the host range of the family Picornaviridae. Arch. Virol. 2015, 160, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Farkas, S.L.; Ihasz, K.; Feher, E.; Bartha, D.; Jakab, F.; Gal, J.; Banyai, K.; Marschang, R.E. Sequencing and phylogenetic analysis identifies candidate members of a new picornavirus genus in terrestrial tortoise species. Arch. Virol. 2015, 160, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Stohr, A.C.; Lopez-Bueno, A.; Blahak, S.; Caeiro, M.F.; Rosa, G.M.; Alves de Matos, A.P.; Martel, A.; Alejo, A.; Marschang, R.E. Phylogeny and differentiation of reptilian and amphibian ranaviruses detected in Europe. PLoS ONE 2015, 10, e0118633. [Google Scholar] [CrossRef] [PubMed]
- Gandar, F.; Wilkie, G.S.; Gatherer, D.; Kerr, K.; Marlier, D.; Diez, M.; Marschang, R.E.; Mast, J.; Dewals, B.G.; Davison, A.J.; et al. The Genome of a Tortoise Herpesvirus (Testudinid Herpesvirus 3) Has a Novel Structure and Contains a Large Region That Is Not Required for Replication In Vitro or Virulence In Vivo. J. Virol. 2015, 89, 11438–11456. [Google Scholar] [CrossRef]
- Schumacher, V.L.; Innis, C.J.; Garner, M.M.; Risatti, G.R.; Nordhausen, R.W.; Gilbert-Marcheterre, K.; Wellehan, J.F., Jr.; Childress, A.L.; Frasca, S., Jr. Sulawesi tortoise adenovirus-1 in two impressed tortoises (Manouria impressa) and a Burmese star tortoise (Geochelone platynota). J. Zoo Wildl. Med. 2012, 43, 501–510. [Google Scholar] [CrossRef]
- Rivera, S.; Wellehan, J.F., Jr.; McManamon, R.; Innis, C.J.; Garner, M.M.; Raphael, B.L.; Gregory, C.R.; Latimer, K.S.; Rodriguez, C.E.; Diaz-Figueroa, O.; et al. Systemic adenovirus infection in Sulawesi tortoises (Indotestudo forsteni) caused by a novel siadenovirus. J. Vet. Diagn. Invest. 2009, 21, 415–426. [Google Scholar] [CrossRef]
- Garcia-Morante, B.; Penzes, J.J.; Costa, T.; Martorell, J.; Martinez, J. Hyperplastic stomatitis and esophagitis in a tortoise (Testudo graeca) associated with an adenovirus infection. J. Vet. Diagn. Invest. 2016, 28, 579–583. [Google Scholar] [CrossRef]
- Doszpoly, A.; Wellehan, J.F., Jr.; Childress, A.L.; Tarjan, Z.L.; Kovacs, E.R.; Harrach, B.; Benko, M. Partial characterization of a new adenovirus lineage discovered in testudinoid turtles. Infect. Genet. Evol. 2013, 17, 106–112. [Google Scholar] [CrossRef]
- Papp, T.; Seybold, J.; Marschang, R.E. Paramyxovirus Infection in a Leopard Tortoise Geochelone pardalis babcocki) with Respiratory Disease. J. Herpetol. Med. Surg. 2010, 20, 64–68. [Google Scholar] [CrossRef]
- Marschang, R.E.; Papp, T.; Frost, J.W. Comparison of paramyxovirus isolates from snakes, lizards and a tortoise. Virus Res. 2009, 144, 272–279. [Google Scholar] [CrossRef]
- Svet-Moldavsky, G.J.; Trubcheninova, L.; Ravkina, L.I. Sarcomas in reptiles induced with Rous virus. Folia Biol. (Praha) 1967, 13, 84. [Google Scholar] [PubMed]
- Varsani, A.; Krupovic, M. Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol. 2017, 3, vew037. [Google Scholar] [CrossRef] [PubMed]
- Cherwa, J.E.J.; Fane, B.A. Microviridae. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 385–393. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Dayaram, A.; Kraberger, S.; Goldstien, S.; Varsani, A.; Krupovic, M. Evolutionary history of ssDNA bacilladnaviruses features horizontal acquisition of the capsid gene from ssRNA nodaviruses. Virology 2017, 504, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Delwart, E.; Rosario, K.; Segales, J.; Varsani, A.; Ictv Report, C. ICTV Virus Taxonomy Profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef]
- Zerbini, F.M.; Briddon, R.W.; Idris, A.; Martin, D.P.; Moriones, E.; Navas-Castillo, J.; Rivera-Bustamante, R.; Roumagnac, P.; Varsani, A.; Ictv Report, C. ICTV Virus Taxonomy Profile: Geminiviridae. J. Gen. Virol. 2017, 98, 131–133. [Google Scholar] [CrossRef]
- Vetten, H.J.; Dale, J.L.; Grigoras, I.; Gronenborn, B.; Harding, R.; Randles, J.W.; Sano, Y.; Thomas, J.E.; Timchenko, T.; Yeh, H.H. Nanoviridae. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 395–404. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Correction to: Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 3213–3214. [Google Scholar] [CrossRef]
- Steel, O.; Kraberger, S.; Sikorski, A.; Young, L.M.; Catchpole, R.J.; Stevens, A.J.; Ladley, J.J.; Coray, D.S.; Stainton, D.; Dayaram, A.; et al. Circular replication-associated protein encoding DNA viruses identified in the faecal matter of various animals in New Zealand. Infect. Genet. Evol. 2016, 43, 151–164. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.(LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Fontenele, R.S.; Lacorte, C.; Lamas, N.S.; Schmidlin, K.; Varsani, A.; Ribeiro, S.G. Single Stranded DNA Viruses Associated with Capybara Faeces Sampled in Brazil. Viruses 2019, 11, 710. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 2015, 1854, 1019–1037. [Google Scholar] [CrossRef]
- Zallot, R.; Oberg, N.O.; Gerlt, J.A. ‘Democratized’ genomic enzymology web tools for functional assignment. Curr. Opin. Chem. Biol. 2018, 47, 77–85. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Stover, B.C.; Muller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef]
- Pei, J.; Grishin, N.V. PROMALS3D: Multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol. Biol. 2014, 1079, 263–271. [Google Scholar] [CrossRef]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Varsani, A.; Koonin, E.V.; Krupovic, M. Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat. Commun. 2019, 10, 3425. [Google Scholar] [CrossRef]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef]
- Krupovic, M.; Ghabrial, S.A.; Jiang, D.; Varsani, A. Genomoviridae: A new family of widespread single-stranded DNA viruses. Arch. Virol. 2016, 161, 2633–2643. [Google Scholar] [CrossRef]
- Yu, X.; Li, B.; Fu, Y.; Jiang, D.; Ghabrial, S.A.; Li, G.; Peng, Y.; Xie, J.; Cheng, J.; Huang, J.; et al. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. USA 2010, 107, 8387–8392. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Visnovsky, G.A.; van Toor, R.F.; Male, M.F.; Waits, K.; Fontenele, R.S.; Varsani, A. Genome Sequences of Two Single-Stranded DNA Viruses Identified in Varroa destructor. Genome Announc. 2018, 6, e00107-18. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, K.; Sepp, T.; Khalifeh, A.; Smith, K.; Fontenele, R.S.; McGraw, K.J.; Varsani, A. Diverse genomoviruses representing eight new and one known species identified in feces and nests of house finches (Haemorhous mexicanus). Arch. Virol. 2019, 164, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.; Rosario, K.; Baker, C.C.M.; Breitbart, M. Discovery of Four Novel Circular Single-Stranded DNA Viruses in Fungus-Farming Termites. Genome Announc. 2018, 6, e00318-18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Harkonen, T.; Hamalainen, A.M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [PubMed]
- Pearson, V.M.; Caudle, S.B.; Rokyta, D.R. Viral recombination blurs taxonomic lines: Examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 2016, 4, e2585. [Google Scholar] [CrossRef] [PubMed]
- Belfort, M. Bacteriophage introns: Parasites within parasites? Trends Genet. 1989, 5, 209–213. [Google Scholar] [CrossRef]
- Creasy, A.; Rosario, K.; Leigh, B.A.; Dishaw, L.J.; Breitbart, M. Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta). Viruses 2018, 10, 404. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rolling Circle Replication (RCR) Endonuclease | Superfamily 3 (SF3) Helicase Motifs | ||||||
|---|---|---|---|---|---|---|---|
| Genus/Group | Accession # | Motif I | Motif II | Motif III | Walker A | Walker B | Motif C |
| Gemykolovirus | MK570209 | MLTYSD | PHFHC | RRWDYVGK | GATRLGKTVWAR | VFDDI | WLCN |
| MK570213 | FLTYSN | PHFHC | RRWDYVGK | GETRLGKTVWAR | IFDDI | WICN | |
| Gemykibivirus | MK570202 | LLTYPQ | VHLHA | KGAAYAIK | GGTRLGKTLWAR | VFDDM | YISN |
| MK570214 | LLTYPQ | VHLHA | KGAAYAIK | GGTRLGKTLWAR | IFDDM | YISN | |
| MK570215 | LLTYPQ | VHLHA | KGAAYAIK | GGTRLGKTLWAR | IFDDM | YISN | |
| MK570205 | LLTYPQ | IHLHA | KGYAYAIK | GPTRLGKTLWAR | VFDDM | YISN | |
| MK570211 | LLTYPQ | VHLHA | KGYAYAIK | GPTRLGKTLWAR | VFDDM | YISN | |
| MK570216 | LLTYPQ | VHLHA | KGYAYAVK | GPTRLGKTLWAR | VFDDM | YISN | |
| MK570207 | LLTYPQ | IHLHA | KGYAYATK | GPTRLGKTLWAR | VFDDM | YISN | |
| MK570208 | LLTYPQ | VHLHA | KGYAYAIK | GPTRLGKTLWAR | VFDDM | YISN | |
| MK570201 | LLTYPQ | IHLHA | KGYAYAIK | GPTRLGKTLWAR | IFDDM | YISN | |
| MK570203 | LLTYPQ | VHLHA | KGYAYAIK | GPTRLGKTLWAR | IFDDM | YISN | |
| MF373640 | LLTYPQ | VHLHA | KGYAYAIK | GPTRLGKTLWAR | VFDDM | YISN | |
| MK570206 | LLTYPQ | VHLHA | KGYAYAIK | GPTRLGKTLWAR | VFDDM | YISN | |
| MF373641 | LLTYPQ | VHLHA | KGYAYAIK | GPTRLGKTLWAR | VFDDM | YISN | |
| MF373638 | LLTYPQ | VHLHA | KGYAYAIK | GPTRLGKTLWAR | VFDDM | YISN | |
| MF373639 | LLTYPQ | VHLHA | KGYAYAVK | GPTRLGKTLWAR | VFDDM | YISN | |
| Gemycircularvirus | MK570218 | LITYAQ | VHLHA | KGYDYAIK | GDSQLGKTVWAR | IFDDM | WLCN |
| MK570204 | LLTYPQ | IHLHA | KGYDYAIK | GDSQLGKTVWAR | VFDDM | WLCN | |
| MK570212 | LLTYPQ | FHLHA | KGYDYAIK | GDSQLGKTLWAR | VFDDM | WLCN | |
| MK570210 | LLTYAQ | IHLHV | TMYDYAIK | GESRLGKTVWAR | VFDDM | WLAN | |
| MK570223 | LFTYAQ | IHFHV | TAYDYACK | GPYGCGKTVWAR | IFDDW | WLCN | |
| MK570217 | LITYSQ | VHLHC | KGWDYACK | GASQTGKTLWAR | VFDDI | WLSN | |
| MK570222 | LITYSQ | IHLHC | KGWDYACK | GASQTGKTLWAR | VFDDI | WLSN | |
| MK570219 | LITYSQ | IHLHC | KGYDYAIK | GASQTGKTLWAR | VFDDI | WLSN | |
| MK570220 | LITYSQ | IHLHC | KGYDYAIK | GASQTGKTLWAR | VFDDI | WLSN | |
| MK570221 | LLTYAQ | SHLHC | AGFDYACK | GEPLTGKTDWAR | IFDDI | WCAN | |
| Unclassified CRESS DNA viruses | MK858252 | FLTYPQ | DHLHA | DVYNYVIK | GPSKTGKTQWAR | VIDDM | ILCN |
| MK858253 | FLTYPQ | DHLHA | DVYNYVIK | GPSKTGKTQWAR | VIDDM | ILCN | |
| MK858254 | FLTYPQ | DHLHA | DVYNYVTK | GPSKTGKTAWAR | VIDDM | ILCN | |
| MK858255 | FLTYPQ | NHLHV | DVYAYITK | GASKTGKTQWAR | VLDDL | ILCN | |
| MK858256 | FLTYPR | DHLHV | HVYRYVRK | GPSKTGKTEWAR | IFDDL | ILCN | |
| MK858257 | FLTFAR | DHRHV | GARQYTQK | GPSKTGKTHWAR | VFDDL | ILCN | |
| MF373642 | FLTYPQ | PHLHC | AVRRYCSK | GNTETGKTTLAK | ILDDM | ITSN | |
| MK858265 | LVTWSQ | LHYHA | DALAYVKK | GPTGSGKTRCAI | IFDDM | - | |
| MK858262 | CKKYRR | PHIQG | ECVTYCKK | GPSGVGKTREVE | VFDDF | RITN | |
| MK858258 | - | - | - | GGSNTGKTTYLR | WIDEF | TLKN | |
| Rep-encoding circular molecules | MK858259 | CFTWNN | PHIQG | DNFKYCTK | GPAGTGKTTWGR | CIEDY | VTSN |
| MK858260 | CFTWNN | PHIQG | DNFKYCTK | GPAGTGKTTWGR | CIEDY | VTSN | |
| MK858263 | LLTFNN | YHTHL | ENRAYVLK | GETGTGKTSSVM | LFDEF | LVSN | |
| MK858264 | - | YHTHL | ENREYIRK | GSTGTGKTSYVM | LFDEF | IISN | |
| Genomoviruses from This Study | Other Genomoviruses | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Query | Genome | Rep | CP | Genome | Rep | CP | |||||||
| Genus | Accession | % ID | Accession # | % ID | Accession # | % ID | Accession # | % ID | Accession # | % ID | Accession # | % ID | Accession # |
| Gemykolovirus | MK570209 | 65.21 | MK570213 | 69.97 | MK570213 | 45.45 | MK570221 | 70.08 | MK939374 | 81.38 | MK939374 | 49.16 | KT862242 |
| MK570213 | 65.21 | MK570209 | 69.97 | MK570209 | 44.44 | MK570223 | 66.96 | MH545501 | 71.26 | MK939374 | 47.42 | MH545501 | |
| Gemykibivirus | MF373638 | 99.82 | MF373641 | 99.69 | MF373641 | 100.00 | MF373641 | 99.22 | MK249293 | 98.46 | MK249293 | 99.67 | MK249293 |
| MF373639 | 99.73 | MF373641 | 99.69 | MF373641 | 99.67 | MF373640 | 99.31 | MK249293 | 98.46 | MK249293 | 100.00 | MK249293 | |
| MF373640 | 96.97 | MF373638 | 97.23 | MF373641 | 100.00 | MF373641 | 96.87 | MK249293 | 97.23 | MK249293 | 99.67 | MK249293 | |
| MF373641 | 99.82 | MF373638 | 99.69 | MF373638 | 100.00 | MF373638 | 99.22 | MK249293 | 98.77 | MK249293 | 99.67 | MK249293 | |
| MK570201 | 96.78 | MF373639 | 98.77 | MK570203 | 99.67 | MF373639 | 96.84 | MK249293 | 94.98 | MK947374 | 99.67 | MK249293 | |
| MK570202 | 99.47 | MK570214 | 99.08 | MK570214 | 100.00 | MK570214 | 90.70 | MK249269 | 83.69 | MK249269 | 98.38 | MK249239 | |
| MK570203 | 94.34 | MK570201 | 98.77 | MK570201 | 98.36 | MK570208 | 95.39 | MK249300 | 95.32 | MK947374 | 99.34 | MK249300 | |
| MK570205 | 95.57 | MK570216 | 95.08 | MK570211 | 99.68 | MK570215 | 97.41 | MK249307 | 99.08 | MK249307 | 98.70 | MK249239 | |
| MK570206 | 96.55 | MF373639 | 99.38 | MF373641 | 99.67 | MK570211 | 91.00 | MK947374 | 98.77 | MK249293 | 98.36 | MK249293 | |
| MK570207 | 97.47 | MK570208 | 97.23 | MK570208 | 97.94 | MK570208 | 92.44 | MK249293 | 96.92 | MK249293 | 97.25 | MK249300 | |
| MK570208 | 97.47 | MK570207 | 97.23 | MK570207 | 98.36 | MK570203 | 92.94 | MK249293 | 96.92 | MK249293 | 98.36 | MK249300 | |
| MK570211 | 94.98 | MK570206 | 95.08 | MK570205 | 99.67 | MK570206 | 92.59 | MK249293 | 97.23 | MK249269 | 98.03 | MK249293 | |
| MK570214 | 99.47 | MK570202 | 99.69 | MK570215 | 100.00 | MK570202 | 90.62 | MK249269 | 83.38 | MK249269 | 98.38 | MK249239 | |
| MK570215 | 98.67 | MK570214 | 99.69 | MK570214 | 100.00 | MK570216 | 90.35 | MK249269 | 83.08 | MK249269 | 99.03 | MK249239 | |
| MK570216 | 95.57 | MK570205 | 94.77 | MF373639 | 100.00 | MK570215 | 94.14 | MK249307 | 94.46 | MK249236 | 99.03 | MK249239 | |
| Gemycircularvirus | MK570204 | 81.28 | MK570212 | 93.99 | MK570212 | 61.76 | MK570210 | 73.52 | MK947372 | 82.53 | MK947372 | 70.10 | MG641202 |
| MK570210 | 67.50 | MK570204 | 58.66 | MK570212 | 61.76 | MK570204 | 76.02 | MK939384 | 92.42 | MK939384 | 75.40 | JQ412056 | |
| MK570212 | 81.28 | MK570204 | 93.99 | MK570204 | 59.28 | MK570218 | 74.28 | MK947372 | 82.83 | MK947372 | 69.81 | MG571096 | |
| MK570217 | 97.56 | MK570222 | 94.80 | MK570222 | 99.02 | MK570222 | 68.13 | KM821747 | 76.76 | KJ547638 | 50.00 | KM510192 | |
| MK570218 | 80.78 | MK570212 | 87.39 | MK570204 | 59.28 | MK570212 | 74.01 | MK947372 | 80.72 | MK947372 | 63.61 | MG571100 | |
| MK570219 | 97.12 | MK570220 | 98.78 | MK570220 | 99.03 | MK570220 | 68.06 | KM821747 | 74.31 | KJ547638 | 53.87 | MK939446 | |
| MK570220 | 97.12 | MK570219 | 98.78 | MK570219 | 99.03 | MK570219 | 67.97 | MK939432 | 74.01 | KJ547638 | 55.08 | MK939446 | |
| MK570221 | 66.65 | MK570204 | 52.00 | MK570210 | 50.33 | MK570204 | 65.90 | MK939384 | 66.03 | MG641197 | 58.36 | KT732806 | |
| MK570222 | 97.56 | MK570217 | 96.94 | MK570219 | 99.02 | MK570217 | 67.91 | KM821747 | 74.62 | KM821747 | 50.43 | KM510192 | |
| MK570223 | 61.64 | MK570210 | 50.76 | MK570210 | 45.54 | MK570204 | 82.49 | MG571087 | 94.22 | MG571087 | 70.39 | KF413620 | |
| Event | Begin | End | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | p-Value | Method |
|---|---|---|---|---|---|---|---|
| I | 190 | 1017 | MK570210 | Unknown | MH939384 | 2.71 × 10−25 | MCS |
| II | 249 * | 1050 | MK570204, MK570212, MK570218 | Unknown | MK570204 | 6.55 × 10−16 | GBMCST |
| III | 243 * | 1009 | MK570218 | Unknown | MK570204 | 5.10 × 10−14 | GBMCT |
| IV | 1539 * | 1591 | MK570222, MK570217 | MF173067 | MK570220, MK570219 | 7.12 × 10−8 | RGBT |
| V | 23 | 1144 | MK570202, MK570214, MK570215 | MK249302, MK249239, MK249243, MK249246, MK249252, MK249256, MK249268, MK249269, MK249275, MK249287, MK249288, MK249307, MK570205, MK570216 | Unknown | 1.67 × 10−52 | RGBMCST |
| VI | 1275 | 2192 | MK570211 | MK249239, MK249243, MK249246, MK249252, MK249256, MK249268, MK249269, MK249275, MK249287, MK249288, MK249302, MK249307 | MF373638, MF373639, MF373640, MF373641, MK249293, MK570206 | 3.64 × 10−86 | GBMCST |
| VII | 333 | 1113 | MF373638, MF373639, MF373640, MF373641, MK570201, MK570203, MK570206, MK570207, MK570208, MK570211 | MK483084 | MH973737, MH973738, MH973739, MH973740, MK249236, MK249241, MK249251, MK249255, MK249271, MK249276, MK249279, MK249286, MK249301 | 7.69 × 10−30 | GBMCST |
| VIII | 1643 | 1958 | MK570216 | MH973737, MH973738, MH973739, MH973740, MK249236, MK249241, MK249251, MK249255, MK249271, MK249276, MK249279, MK249286, MK249293, MK249301, MK570206 | MK249239, MK249243, MK249246, MK249252, MK249256, MK249268, MK249269, MK249275, MK249287, MK249288, MK249302, MK249307, MK570205 | 5.32 × 10−25 | RGBMCST |
| IX | 1993 | 2211 | MK570205, MK570216 | MF373638, MF373639, MF373640, MF373641, MH973737, MH973738, MH973739, MH973740, MK249236, MK249241, MK249251, MK249255, MK249271, MK249276, MK249279, MK249286, MK249293, MK249296, MK249301, MK570206, MK947374 | MK249239, MK249243, MK249246, MK249252, MK249256, MK249268, MK249269, MK249275, MK249287, MK249288, MK249302 | 7.83 × 10−22 | GBMCST |
| Unclassified CRESS DNA Viruses from This Study | Other Unclassified CRESS DNA Viruses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Query | Rep | CP | Rep | CP | |||||
| Virus Group/Molecules | Accession # | Accession # | %ID | Accession # | %ID | Accession # | %ID | Accession # | %ID |
| Unclassified CRESS DNA virus | MF373642 | MK858254 | 29.76 | - | - | KY487833 | 42.00 | - | - |
| MK858252 | MK858253 | 100.00 | MK858253 | 99.00 | MK570182 | 78.42 | MK858254 | 38.68 | |
| MK858253 | MK858252 | 100.00 | MK858253 | 99.00 | MK570182 | 78.42 | MK858254 | 38.68 | |
| MK858254 | MK858252 | 81.16 | MK858255 | 39.16 | MK570182 | 75.99 | MK570182 | 39.25 | |
| MK858255 | MK858252 | 79.03 | MK858254 | 39.25 | MK570182 | 83.57 | MK570165 | 53.64 | |
| MK858256 | MK858255 | 65.75 | MK858257 | 33.47 | MK570182 | 64.44 | MK570168 | 33.46 | |
| MK858257 | MK858256 | 56.23 | MK858254 | 25.21 | MK570165 | 54.08 | MK570168 | 42.45 | |
| MK858258 | MK858260 | 35.57 | MK858261 | 52.65 | MK570170 | 45.86 | - | - | |
| MK858262 | MK858265 | 30.80 | MH616676 | 46.77 | KY302869 | 28.12 | |||
| MK858265 | MK858262 | 30.80 | - | - | KM821751 | 30.80 | - | - | |
| Circular molecule | MK858259 | MK858260 | 99.70 | - | - | MF118167 | 49.34 | - | - |
| MK858260 | MK858259 | 99.70 | - | - | MF118167 | 49.34 | - | - | |
| MK858261 | - | - | MK858258 | 52.65 | - | - | - | - | |
| MK858263 | MK858264 | 72.53 | - | - | MH616676 | 47.17 | - | - | |
| MK858264 | MK858263 | 72.53 | - | - | MH616676 | 47.17 | - | - | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, 143. https://doi.org/10.3390/v12020143
Orton JP, Morales M, Fontenele RS, Schmidlin K, Kraberger S, Leavitt DJ, Webster TH, Wilson MA, Kusumi K, Dolby GA, et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses. 2020; 12(2):143. https://doi.org/10.3390/v12020143
Chicago/Turabian StyleOrton, Joseph P., Matheo Morales, Rafaela S. Fontenele, Kara Schmidlin, Simona Kraberger, Daniel J. Leavitt, Timothy H. Webster, Melissa A. Wilson, Kenro Kusumi, Greer A. Dolby, and et al. 2020. "Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses" Viruses 12, no. 2: 143. https://doi.org/10.3390/v12020143
APA StyleOrton, J. P., Morales, M., Fontenele, R. S., Schmidlin, K., Kraberger, S., Leavitt, D. J., Webster, T. H., Wilson, M. A., Kusumi, K., Dolby, G. A., & Varsani, A. (2020). Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses, 12(2), 143. https://doi.org/10.3390/v12020143