Virus Metagenomics in Farm Animals: A Systematic Review



Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy and Selection Criteria
2.2. Data Extraction and Analysis
3. Results
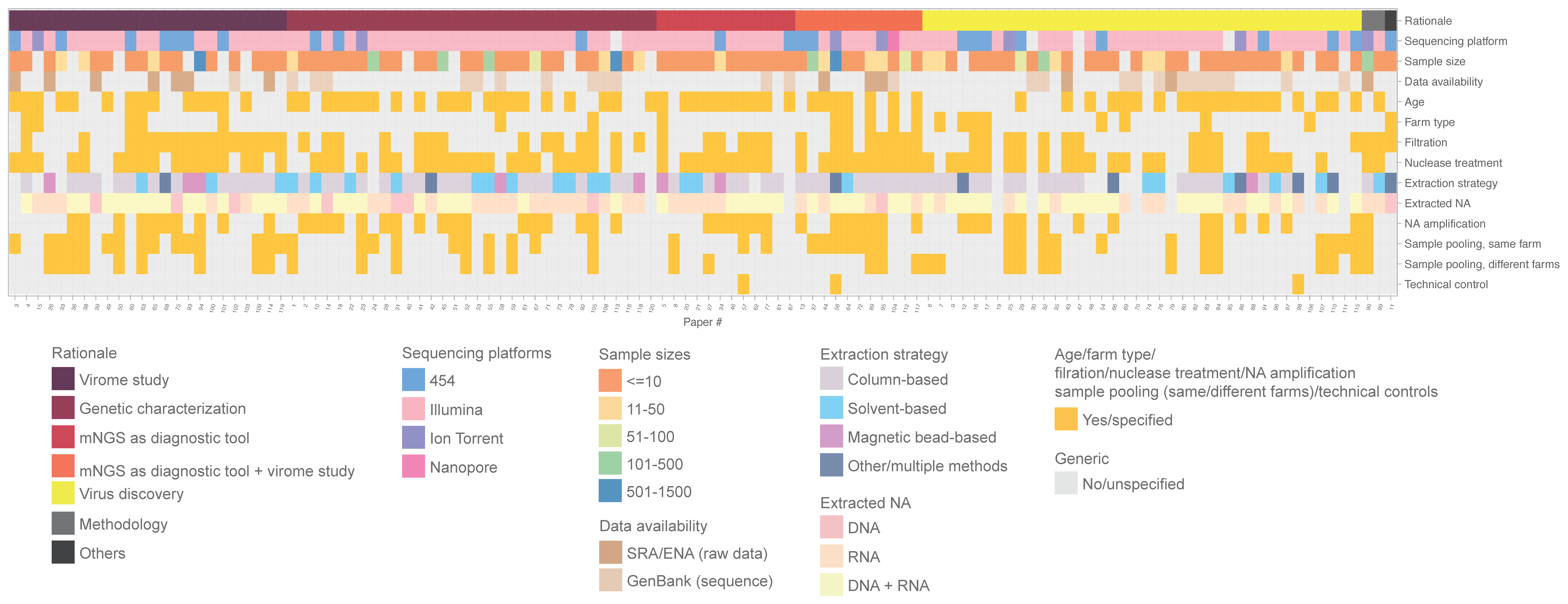
3.1. Overall Descriptions of All Studies
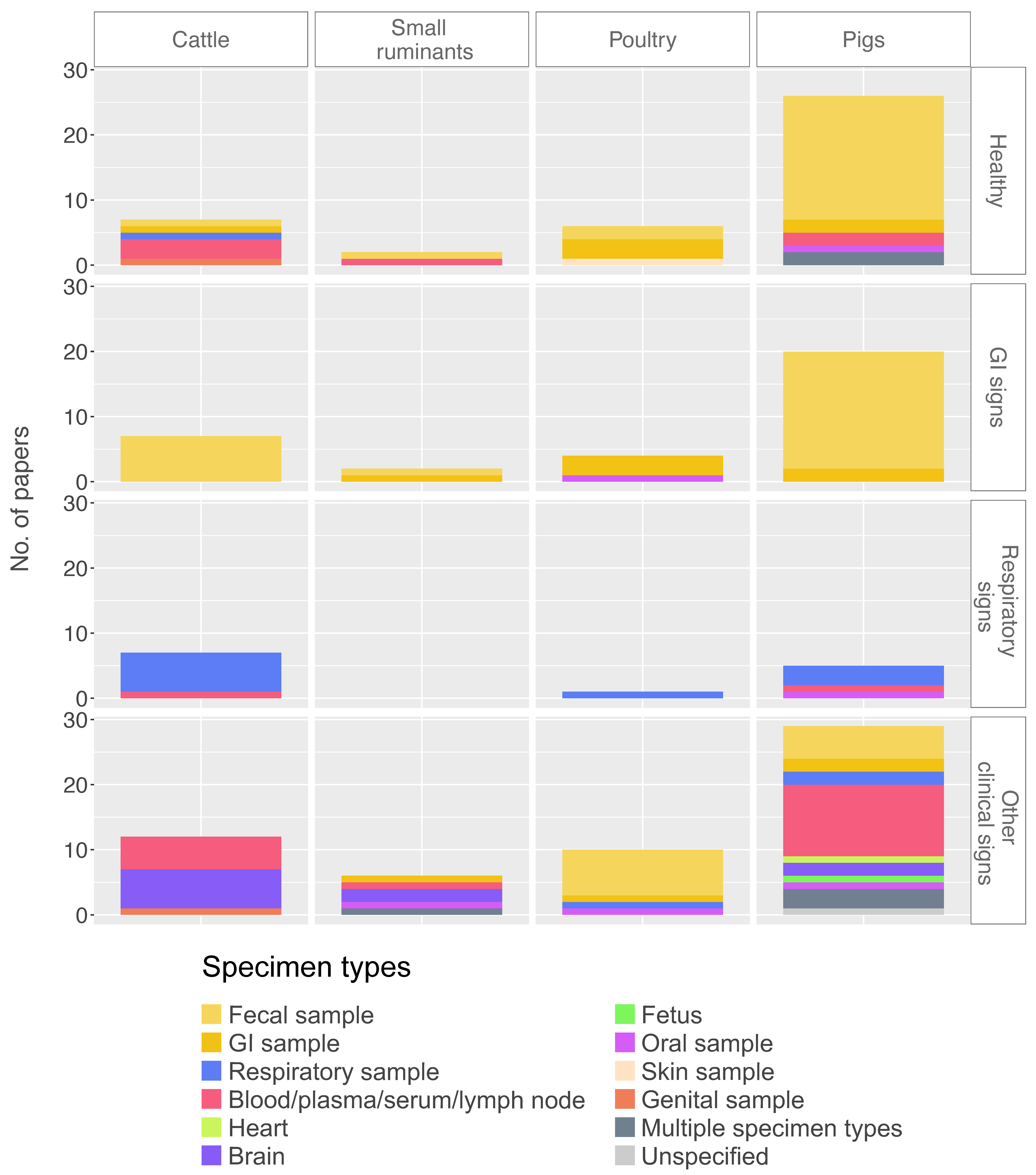
3.2. Farm Animals, Health Conditions, and Specimen Types
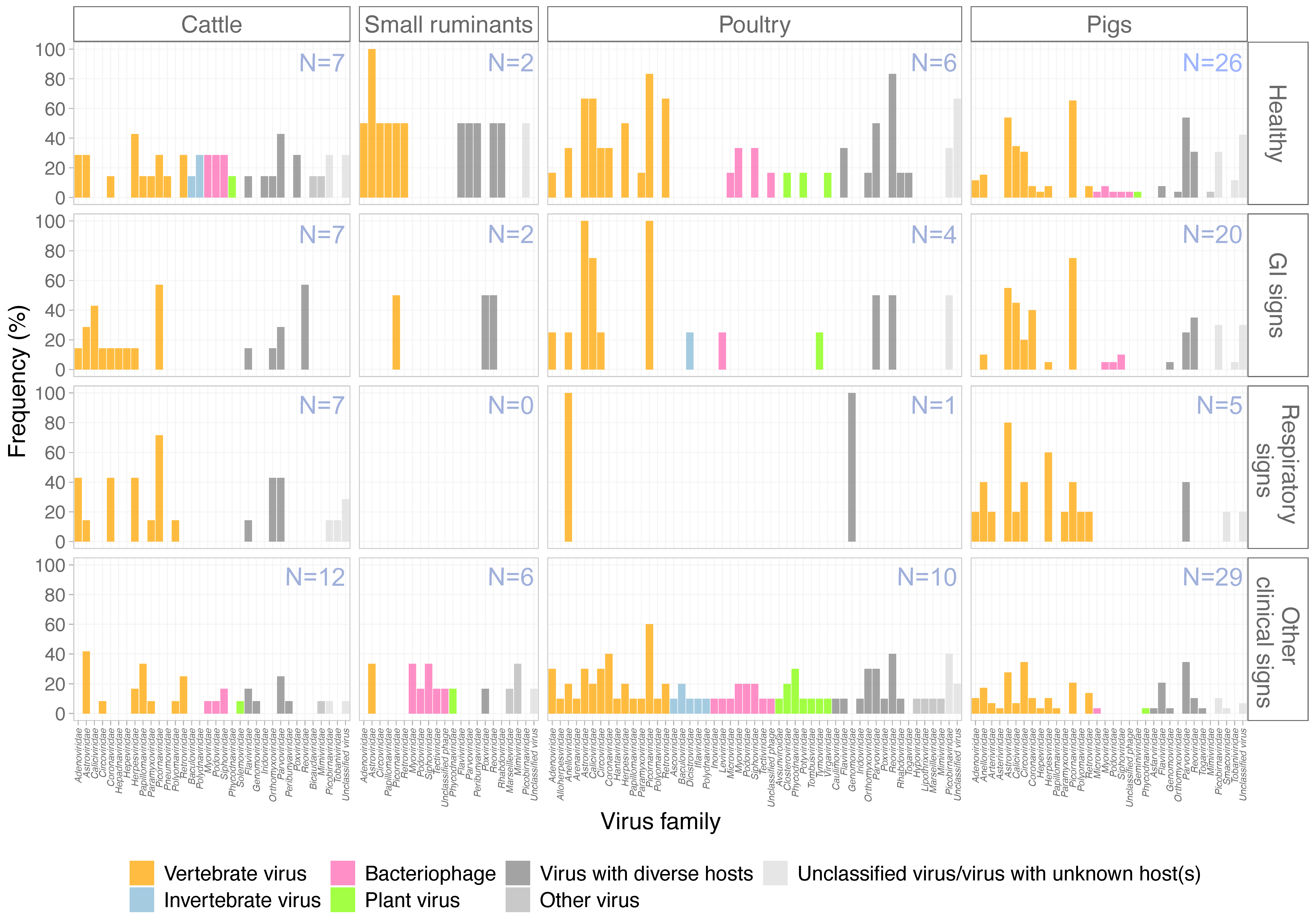
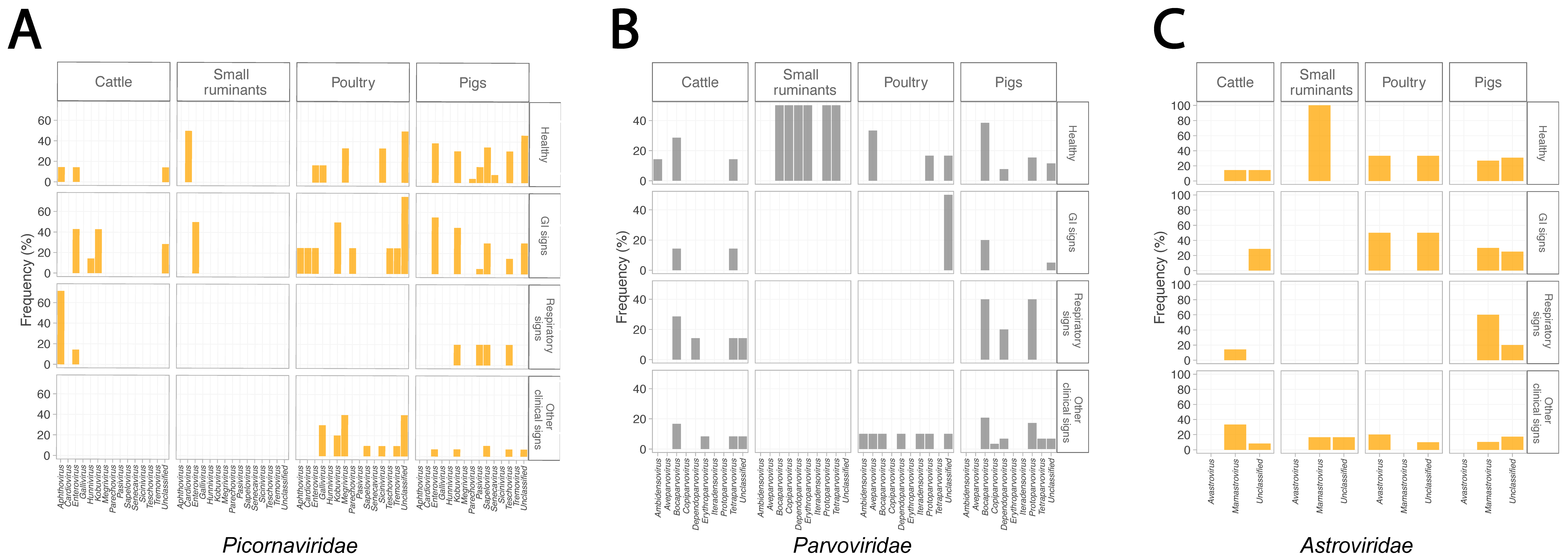
3.3. Virus Diversity in Different Farm Animals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Wolfe, N.D.; Dunavan, C.P.; Diamond, J. Origins of major human infectious diseases. Nature 2007, 447, 279–283. [Google Scholar] [CrossRef]
- Jones, B.A.; Grace, D.; Kock, R.; Alonso, S.; Rushton, J.; Said, M.Y.; McKeever, D.; Mutua, F.; Young, J.; McDermott, J.; et al. Zoonosis emergence linked to agricultural intensification and environmental change. Proc. Natl. Acad. Sci. USA 2013, 110, 8399–8404. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Hsu, V.P.; Hossain, M.J.; Parashar, U.D.; Ali, M.M.; Ksiazek, T.G.; Kuzmin, I.; Niezgoda, M.; Rupprecht, C.; Bresee, J.; Breiman, R.F. Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis. 2004, 10, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P. Nipah virus outbreak in India. Lancet 2018, 391, 2200. [Google Scholar] [CrossRef]
- Roberts, L. Nigeria hit by unprecedented Lassa fever outbreak. Science 2018, 359, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.Y.; Zhou, B.; Wang, J.; Chai, Y.; Shen, Y.; Chen, X.; Ma, C.; Hong, W.; Chen, Y.; Zhang, Y.; et al. Dissemination, divergence and establishment of H7N9 influenza viruses in China. Nature 2015, 522, 102–105. [Google Scholar] [CrossRef]
- Tambo, E.; Olalubi, O.A.; Sacko, M. Rift valley fever epidemic in Niger near border with Mali. Lancet Infect. Dis. 2016, 16, 1319–1320. [Google Scholar] [CrossRef]
- Fraaij, P.L.A.; Wildschut, E.D.; Houmes, R.J.; Swaan, C.M.; Hoebe, C.J.; de Jonge, H.C.C.; Tolsma, P.; de Kleer, I.; Pas, S.D.; Oude Munnink, B.B.; et al. Severe acute respiratory infection caused by swine influenza virus in a child necessitating extracorporeal membrane oxygenation (ECMO), The Netherlands, October 2016. Eurosurveillance 2016, 21, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Coker, R.; Rushton, J.; Mounier-Jack, S.; Karimuribo, E.; Lutumba, P.; Kambarage, D.; Pfeiffer, D.U.; Stärk, K.; Rweyemamu, M. Towards a conceptual framework to support one-health research for policy on emerging zoonoses. Lancet Infect. Dis. 2011, 11, 326–331. [Google Scholar] [CrossRef]
- Castillo-Chavez, C.; Curtiss, R.; Daszak, P.; Levin, S.A.; Patterson-Lomba, O.; Perrings, C.; Poste, G.; Towers, S. Beyond Ebola: Lessons to mitigate future pandemics. Lancet Glob. Health 2015, 3, e354–e355. [Google Scholar] [CrossRef]
- Koopmans, M.; Wilbrink, B.; Conyn, M.; Natrop, G.; Van Der Nat, H.; Vennema, H.; Meijer, A.; Van Steenbergen, J.; Fouchier, R.; Osterhaus, A.; et al. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet 2004, 363, 587–593. [Google Scholar] [CrossRef]
- Sims, L.D.; Domenech, J.; Benigno, C.; Kahn, S.; Kamata, A.; Lubroth, J.; Martin, V.; Roeder, P. Origin and evolution of highly pathogenic H5N1 avian influenza in Asia. Vet. Rec. 2005, 157, 159–164. [Google Scholar] [CrossRef]
- Van der Hoek, W.; Dijkstra, F.; Schimmer, B.; Schneeberger, P.M.; Vellema, P.; Wijkmans, C.; ter Schegget, R.; Hackert, V.; van Duynhoven, Y. Q fever in the Netherlands: An update on the epidemiology and control measures. Eurosurveillance 2010, 15, 15. [Google Scholar]
- Schimmer, B.; Dijkstra, F.; Vellema, P.; Schneeberger, P.M.; Hackert, V.; ter Schegget, R.; Wijkmans, C.; van Duynhoven, Y.; van der Hoek, W. Sustained intensive transmission of Q fever in the south of the Netherlands, 2009. Eurosurveillance 2009, 14, 19210. [Google Scholar] [CrossRef]
- Destoumieux-Garzón, D.; Mavingui, P.; Boetsch, G.; Boissier, J.; Darriet, F.; Duboz, P.; Fritsch, C.; Giraudoux, P.; Le Roux, F.; Morand, S.; et al. The one health concept: 10 years old and a long road ahead. Front. Vet. Sci. 2018, 5, 14. [Google Scholar] [CrossRef]
- Quan, P.L.; Wagner, T.A.; Briese, T.; Torgerson, T.R.; Hornig, M.; Tashmukhamedova, A.; Firth, C.; Palacios, G.; Baisre-de-Leon, A.; Paddock, C.D.; et al. Astrovirus encephalitis in boy with X-linked agammaglobulinemia. Emerg. Infect. Dis. 2010, 16, 918–925. [Google Scholar] [CrossRef]
- Naccache, S.N.; Peggs, K.S.; Mattes, F.M.; Phadke, R.; Garson, J.A.; Grant, P.; Samayoa, E.; Federman, S.; Miller, S.; Lunn, M.P.; et al. Diagnosis of neuroinvasive astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin. Infect. Dis. 2015, 60, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Coffey, L.L.; Murkey, J.; Symmes, K.; Sample, H.A.; Wilson, M.R.; Naccache, S.N.; Arevalo, S.; Somasekar, S.; Federman, S.; et al. Diagnosis of fatal human case of St. Louis encephalitis virus infection by metagenomic sequencing, California. 2016. Emerg. Infect. Dis. 2017, 23, 1694–1698. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef] [PubMed]
- Blomström, A.L.; Fossum, C.; Wallgren, P.; Berg, M. Viral metagenomic analysis displays the Co-infection situation in healthy and PMWS affected pigs. PLoS ONE 2016, 11, e0166863. [Google Scholar] [CrossRef]
- Yu, X.; Jin, T.; Cui, Y.; Pu, X.; Li, J.; Xu, J.; Liu, G.; Jia, H.; Liu, D.; Song, S.; et al. Influenza H7N9 and H9N2 Viruses: Coexistence in Poultry Linked to Human H7N9 Infection and Genome Characteristics. J. Virol. 2014, 88, 3423–3431. [Google Scholar] [CrossRef]
- Nieuwenhuijse, D.F.; Koopmans, M.P.G. Metagenomic sequencing for surveillance of food-and waterborne viral diseases. Front. Microbiol. 2017, 8, 69. [Google Scholar] [CrossRef]
- Yinda, C.K.; Vanhulle, E.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Shi, C.; Ghogomu, S.M.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Gut Virome Analysis of Cameroonians Reveals High Diversity of Enteric Viruses, Including Potential Interspecies Transmitted Viruses. mSphere 2019, 4, e00585-18. [Google Scholar] [CrossRef]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Qiu, M.; Guan, W.J.; Li, J.M.; Chen, S.W.; Cheng, M.J.; Huo, S.T.; Chen, Z.; Wu, Y.; Jiang, L.N.; et al. Viral metagenomics of six bat species in close contact with humans in southern China. Arch. Virol. 2018, 163, 73–88. [Google Scholar] [CrossRef]
- Blomström, A.L.; Ye, X.; Fossum, C.; Wallgren, P.; Berg, M. Characterisation of the virome of tonsils from conventional pigs and from specific pathogen-free pigs. Viruses 2018, 10, 382. [Google Scholar] [CrossRef]
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science 2018, 359, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 5 December 2019).
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. Available online: https://cran.r-project.org/package=dplyr (accessed on 5 December 2019).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. Available online: https://CRAN.R-project.org/package=stringr (accessed on 5 December 2019).
- Hulo, C.; De Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2017, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Ng, T.F.F.; Lager, K.M.; Alt, D.P.; Delwart, E.L.; Pogranichniy, R.M. Unique circovirus-like genome detected in pig feces. Genome Announc. 2014, 2, e00251-14. [Google Scholar] [CrossRef]
- Hause, B.M.; Collin, E.A.; Peddireddi, L.; Yuan, F.; Chen, Z.; Hesse, R.A.; Gauger, P.C.; Clement, T.; Fang, Y.; Anderson, G. Discovery of a novel putative atypical porcine pestivirus in pigs in the USA. J. Gen. Virol. 2015, 96, 2994–2998. [Google Scholar] [CrossRef]
- Pankovics, P.; Boros, Á.; Nemes, C.; Kapusinszky, B.; Delwart, E.; Reuter, G. Molecular characterization of a novel picobirnavirus in a chicken. Arch. Virol. 2018, 163, 3455–3458. [Google Scholar] [CrossRef]
- Yang, C.; Wang, L.; Shen, H.; Zheng, Y.; Bade, S.A.; Gauger, P.C.; Chen, Q.; Zhang, J.; Guo, B.; Yoon, K.J.; et al. Detection and genetic characterization of porcine pegivirus in pigs in the United States. Transbound. Emerg. Dis. 2018, 65, 618–626. [Google Scholar] [CrossRef]
- Mitra, N.; Cernicchiaro, N.; Torres, S.; Li, F.; Hause, B.M. Metagenomic characterization of the virome associated with bovine respiratory disease in feedlot cattle identified novel viruses and suggests an etiologic role for influenza D virus. J. Gen. Virol. 2016, 97, 1771–1784. [Google Scholar] [CrossRef]
- Grierson, S.S.; McGowan, S.; Cook, C.; Steinbach, F.; Choudhury, B. Molecular and in vitro characterisation of hepatitis E virus from UK pigs. Virology 2019, 527, 116–121. [Google Scholar] [CrossRef]
- Hause, B.M.; Duff, J.W.; Scheidt, A.; Anderson, G. Virus detection using metagenomic sequencing of swine nasal and rectal swabs. J. Swine Health Prod. 2016, 24, 304–308. [Google Scholar]
- Chen, X.; Zhang, B.; Yue, H.; Wang, Y.; Zhou, F.; Zhang, Q.; Tang, C. A novel astrovirus species in the gut of yaks with diarrhoea in the Qinghai-Tibetan plateau, 2013. J. Gen. Virol. 2015, 96, 3672–3680. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Kondov, N.O.; Deng, X.; Van Eenennaam, A.; Neibergs, H.L.; Delwart, E. A Metagenomics and Case-Control Study To Identify Viruses Associated with Bovine Respiratory Disease. J. Virol. 2015, 89, 5340–5349. [Google Scholar] [CrossRef]
- Hause, B.M.; Collin, E.A.; Anderson, J.; Hesse, R.A.; Anderson, G. Bovine rhinitis viruses are common in U.S. cattle with bovine respiratory disease. PLoS ONE 2015, 10, e0121998. [Google Scholar] [CrossRef]
- Guo, Z.; He, Q.; Tang, C.; Zhang, B.; Yue, H. Identification and genomic characterization of a novel CRESS DNA virus from a calf with severe hemorrhagic enteritis in China. Virus Res. 2018, 255, 141–146. [Google Scholar] [CrossRef]
- Dumarest, M.; Muth, E.; Cheval, J.; Gratigny, M.; Hébert, C.; Gagnieur, L.; Eloit, M. Viral diversity in swine intestinal mucus used for the manufacture of heparin as analyzed by high-throughput sequencing. Biologicals 2015, 43, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, R.; Sameroff, S.; Hesse, R.A.; Hause, B.M.; Desai, A.; Jain, K.; Ian Lipkin, W. Discovery of a novel nidovirus in cattle with respiratory disease. J. Gen. Virol. 2015, 96, 2188–2193. [Google Scholar] [CrossRef]
- Wang, H.; Li, S.; Mahmood, A.; Yang, S.; Wang, X.; Shen, Q.; Shan, T.; Deng, X.; Li, J.; Hua, X.; et al. Plasma virome of cattle from forest region revealed diverse small circular ssDNA viral genomes. Virol. J. 2018, 15, 11. [Google Scholar] [CrossRef]
- Ling, Y.; Zhang, X.; Qi, G.; Yang, S.; Jingjiao, L.; Shen, Q.; Wang, X.; Cui, L.; Hua, X.; Deng, X.; et al. Viral metagenomics reveals significant viruses in the genital tract of apparently healthy dairy cows. Arch. Virol. 2019, 164, 1059–1067. [Google Scholar] [CrossRef]
- Cibulski, S.P.; Teixeira, T.F.; dos Santos, H.F.; de Sales Lima, F.E.; Scheffer, C.M.; Varela, A.P.M.; de Lima, D.A.; Schmidt, C.; Silveira, F.; de Almeida, L.L.; et al. Ungulate copiparvovirus 1 (bovine parvovirus 2): Characterization of a new genotype and associated viremia in different bovine age groups. Virus Genes 2016, 52, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Kapusinszky, B.; Yugo, D.M.; Phan, T.G.; Deng, X.; Kanevsky, I.; Opriessnig, T.; Woolums, A.R.; Hurley, D.J.; Meng, X.J.; et al. Virome of US bovine calf serum. Biologicals 2017, 46, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Blomström, A.L.; Oma, V.; Khatri, M.; Hansen, H.H.; Stokstad, M.; Berg, M.; Myrmel, M. Genome sequence of a bovine rhinitis B virus identified in cattle in Sweden. Genome Announc. 2017, 5, e00172-17. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Omatsu, T.; Aoki, H.; Otomaru, K.; Uto, T.; Koizumi, M.; Minami-Fukuda, F.; Takai, H.; Murakami, T.; Masuda, T.; et al. Full genome analysis of bovine astrovirus from fecal samples of cattle in Japan: Identification of possible interspecies transmission of bovine astrovirus. Arch. Virol. 2015, 160, 2491–2501. [Google Scholar] [CrossRef]
- Mekata, H.; Yamamoto, M.; Hamabe, S.; Tanaka, H.; Omatsu, T.; Mizutani, T.; Hause, B.M.; Okabayashi, T. Molecular epidemiological survey and phylogenetic analysis of bovine influenza D virus in Japan. Transbound. Emerg. Dis. 2018, 65, e355–e360. [Google Scholar] [CrossRef]
- Ferragut, F.; Vega, C.G.; Mauroy, A.; Conceição-Neto, N.; Zeller, M.; Heylen, E.; Uriarte, E.L.; Bilbao, G.; Bok, M.; Matthijnssens, J.; et al. Molecular detection of bovine Noroviruses in Argentinean dairy calves: Circulation of a tentative new genotype. Infect. Genet. Evol. 2016, 40, 144–150. [Google Scholar] [CrossRef]
- Hayashi-Miyamoto, M.; Murakami, T.; Minami-Fukuda, F.; Tsuchiaka, S.; Kishimoto, M.; Sano, K.; Naoi, Y.; Asano, K.; Ichimaru, T.; Haga, K.; et al. Diversity in VP3, NSP3, and NSP4 of rotavirus B detected from Japanese cattle. Infect. Genet. Evol. 2017, 49, 97–103. [Google Scholar] [CrossRef]
- Omatsu, T.; Tsuchiaka, S.; Hirata, T.; Shiroma, Y.; Okazaki, S.; Katayama, Y.; Oba, M.; Nishiura, N.; Sassa, Y.; Furuya, T.; et al. Detection of enterovirus genome sequence from diarrheal feces of goat. Virus Genes 2014, 48, 550–552. [Google Scholar] [CrossRef]
- Maganga, G.D.; Relmy, A.; Bakkali-Kassimi, L.; Ngoubangoye, B.; Tsoumbou, T.; Bouchier, C.; N’Dilimabaka, N.; Leroy, E.M.; Zientara, S.; Berthet, N. Molecular characterization of Orf virus in goats in Gabon, Central Africa. Virol. J. 2016, 13, 79. [Google Scholar] [CrossRef]
- Chen, F.; Knutson, T.P.; Ciarlet, M.; Sturos, M.; Marthaler, D.G. Complete genome characterization of a rotavirus B (RVB) strain identified in alpine goat kids with enteritis reveals inter-species transmission with RVB bovine strains. J. Gen. Virol. 2018, 99, 457–463. [Google Scholar] [CrossRef]
- Yang, L.E.; Zhao, Z.; Hou, G.; Zhang, C.; Liu, J.; Xu, L.; Li, W.; Tan, Z.; Tu, C.; He, B. Genomes and seroprevalence of severe fever with thrombocytopenia syndrome virus and Nairobi sheep disease virus in Haemaphysalis longicornis ticks and goats in Hubei, China. Virology 2019, 529, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Ballard, L.L.; Duke, M.V.; Scheffler, B.E.; Zsak, L. Metagenomic analysis of the turkey gut RNA virus community. Virol. J. 2010, 7, 313. [Google Scholar] [CrossRef]
- Shah, J.D.; Baller, J.; Zhang, Y.; Silverstein, K.; Xing, Z.; Cardona, C.J. Comparison of tissue sample processing methods for harvesting the viral metagenome and a snapshot of the RNA viral community in a turkey gut. J. Virol. Methods 2014, 209, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Yoon, S.J.; Lee, H.S.; Kwon, Y.K. Identification of a picornavirus from chickens with transmissible viral proventriculitis using metagenomic analysis. Arch. Virol. 2015, 160, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Devaney, R.; Trudgett, J.; Trudgett, A.; Meharg, C.; Smyth, V. A metagenomic comparison of endemic viruses from broiler chickens with runting-stunting syndrome and from normal birds. Avian Pathol. 2016, 45, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Chen, J.M.; Wang, T.; Hou, G.Y.; Zhuang, Q.Y.; Wu, R.; Wang, K.C. Detection of viromes of RNA viruses using the next generation sequencing libraries prepared by three methods. Virus Res. 2017, 237, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Lima, D.A.; Cibulski, S.P.; Tochetto, C.; Varela, A.P.M.; Finkler, F.; Teixeira, T.F.; Loiko, M.R.; Cerva, C.; Junqueira, D.M.; Mayer, F.Q.; et al. The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res. 2019, 261, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lima, D.A.; Cibulski, S.P.; Finkler, F.; Teixeira, T.F.; Varela, A.P.M.; Cerva, C.; Loiko, M.R.; Scheffer, C.M.; Dos Santos, H.F.; Mayer, F.Q.; et al. Faecal virome of healthy chickens reveals a large diversity of the eukaryote viral community, including novel circular ssDNA viruses. J. Gen. Virol. 2017, 98, 690–703. [Google Scholar] [CrossRef]
- Boros, A.; Pankovics, P.; Knowles, N.J.; Nemes, C.; Delwart, E.; Reuter, G. Comparative Complete Genome Analysis of Chicken and Turkey Megriviruses (Family Picornaviridae): Long 3 Untranslated Regions with a Potential Second Open Reading Frame and Evidence for Possible Recombination. J. Virol. 2014, 88, 6434–6443. [Google Scholar] [CrossRef]
- Day, J.M.; Zsak, L. Investigating Turkey Enteric Picornavirus and Its Association with Enteric Disease in Poults. Avian Dis. 2015, 59, 138–142. [Google Scholar] [CrossRef]
- Farlow, J.; Donduashvili, M.; Kokhreidze, M.; Kotorashvili, A.; Vepkhvadze, N.G.; Kotaria, N.; Gulbani, A. Intra-epidemic genome variation in highly pathogenic African swine fever virus (ASFV) from the country of Georgia. Virol. J. 2018, 15, 190. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The Fecal Virome of Pigs on a High-Density Farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef] [PubMed]
- Lager, K.M.; Ng, T.F.; Bayles, D.O.; Alt, D.P.; Delwart, E.L.; Cheung, A.K. Diversity of viruses detected by deep sequencing in pigs from a common background. J. Vet. Diagn. Investig. 2012, 24, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Sachsenröder, J.; Twardziok, S.O.; Scheuch, M.; Johne, R. The general composition of the faecal virome of pigs depends on age, but not on feeding with a probiotic bacterium. PLoS ONE 2014, 9, e88888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tang, C.; Yue, H.; Ren, Y.; Song, Z. Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrhoeic faeces in China. J. Gen. Virol. 2014, 95, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Amimo, J.O.; El Zowalaty, M.E.; Githae, D.; Wamalwa, M.; Djikeng, A.; Nasrallah, G.K. Metagenomic analysis demonstrates the diversity of the fecal virome in asymptomatic pigs in East Africa. Arch. Virol. 2016, 161, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Hargitai, R.; Pankovics, P.; Kertész, A.M.; Bíró, H.; Boros, Á.; Phan, T.G.; Delwart, E.; Reuter, G. Detection and genetic characterization of a novel parvovirus distantly related to human bufavirus in domestic pigs. Arch. Virol. 2016, 161, 1033–1037. [Google Scholar] [CrossRef]
- Cheung, A.K.; Ng, T.F.; Lager, K.M.; Bayles, D.O.; Alt, D.P.; Delwart, E.L.; Pogranichniy, R.M.; Kehrli, M.E. A divergent clade of circular single-stranded DNA viruses from pig feces. Arch. Virol. 2013, 158, 2157–2162. [Google Scholar] [CrossRef]
- Theuns, S.; Conceição-Neto, N.; Zeller, M.; Heylen, E.; Roukaerts, I.D.M.; Desmarets, L.M.B.; Van Ranst, M.; Nauwynck, H.J.; Matthijnssens, J. Characterization of a genetically heterogeneous porcine rotavirus C, and other viruses present in the fecal virome of a non-diarrheic Belgian piglet. Infect. Genet. Evol. 2016, 43, 135–145. [Google Scholar] [CrossRef]
- Akagami, M.; Ito, M.; Niira, K.; Kuroda, M.; Masuda, T.; Haga, K.; Tsuchiaka, S.; Naoi, Y.; Kishimoto, M.; Sano, K.; et al. Complete genome analysis of porcine kobuviruses from the feces of pigs in Japan. Virus Genes 2017, 53, 593–602. [Google Scholar] [CrossRef]
- Conceição-Neto, N.; Theuns, S.; Cui, T.; Zeller, M.; Yinda, C.K.; Christiaens, I.; Heylen, E.; Van Ranst, M.; Carpentier, S.; Nauwynck, H.J.; et al. Identification of an enterovirus recombinant with a torovirus-like gene insertion during a diarrhea outbreak in fattening pigs. Virus Evol. 2017, 3, vex024. [Google Scholar] [CrossRef] [PubMed]
- Knutson, T.P.; Velayudhan, B.T.; Marthaler, D.G. A porcine enterovirus G associated with enteric disease contains a novel papain-like cysteine protease. J. Gen. Virol. 2017, 98, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Masuda, T.; Ito, M.; Naoi, Y.; Doan, Y.H.; Haga, K.; Tsuchiaka, S.; Kishimoto, M.; Sano, K.; Omatsu, T.; et al. Genetic diversity and intergenogroup recombination events of sapoviruses detected from feces of pigs in Japan. Infect. Genet. Evol. 2017, 55, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, L.; Zheng, Y.; Zhang, J.; Guo, B.; Yoon, K.J.; Gauger, P.C.; Harmon, K.M.; Main, R.G.; Li, G. Metagenomic analysis of the RNA fraction of the fecal virome indicates high diversity in pigs infected by porcine endemic diarrhea virus in the United States. Virol. J. 2018, 15, 95. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sunaga, F.; Naoi, Y.; Ito, M.; Takagi, H.; Katayama, Y.; Omatsu, T.; Oba, M.; Sakaguchi, S.; Furuya, T.; et al. Whole genome analysis of a novel picornavirus related to the Enterovirus/Sapelovirus supergroup from porcine feces in Japan. Virus Res. 2018, 257, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Naoi, Y.; Ito, M.; Masuda, T.; Katayama, Y.; Sakaguchi, S.; Omatsu, T.; Furuya, T.; Yamasato, H.; Sunaga, F.; et al. Metagenomic identification and sequence analysis of a Teschovirus A-related virus in porcine feces in Japan, 2014–2016. Infect. Genet. Evol. 2018, 66, 210–216. [Google Scholar] [CrossRef]
- Theuns, S.; Vanmechelen, B.; Bernaert, Q.; Deboutte, W.; Vandenhole, M.; Beller, L.; Matthijnssens, J.; Maes, P.; Nauwynck, H.J. Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Sci. Rep. 2018, 8, 9830. [Google Scholar] [CrossRef]
- Tsuchiaka, S.; Naoi, Y.; Imai, R.; Masuda, T.; Ito, M.; Akagami, M.; Ouchi, Y.; Ishii, K.; Sakaguchi, S.; Omatsu, T.; et al. Genetic diversity and recombination of enterovirus G strains in Japanese pigs: High prevalence of strains carrying a papain-like cysteine protease sequence in the enterovirus G population. PLoS ONE 2018, 13, e0190819. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Liu, Z.; Fu, X.; Yuan, J.; Zhao, J.; Lin, Y.; Shen, Q.; Wang, X.; Deng, X.; et al. Full-length and defective enterovirus G genomes with distinct torovirus protease insertions are highly prevalent on a Chinese pig farm. Arch. Virol. 2018, 163, 2471–2476. [Google Scholar] [CrossRef]
- Hause, B.M.; Padmanabhan, A.; Pedersen, K.; Gidlewski, T. Feral swine virome is dominated by single-stranded DNA viruses and contains a novel Orthopneumovirus which circulates both in feral and domestic swine. J. Gen. Virol. 2016, 97, 2090–2095. [Google Scholar] [CrossRef]
- Qin, S.; Ruan, W.; Yue, H.; Tang, C.; Zhou, K.; Zhang, B. Viral communities associated with porcine respiratory disease complex in intensive commercial farms in Sichuan province, China. Sci. Rep. 2018, 8, 13341. [Google Scholar] [CrossRef]
- Zheng, S.; Wu, X.; Zhang, L.; Xin, C.; Liu, Y.; Shi, J.; Peng, Z.; Xu, S.; Fu, F.; Yu, J.; et al. The occurrence of porcine circovirus 3 without clinical infection signs in Shandong Province. Transbound. Emerg. Dis. 2017, 64, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Tochetto, C.; Lima, D.A.; Varela, A.P.M.; Loiko, M.R.; Paim, W.P.; Scheffer, C.M.; Herpich, J.I.; Cerva, C.; Schmitd, C.; Cibulski, S.P.; et al. Full-Genome Sequence of Porcine Circovirus type 3 recovered from serum of sows with stillbirths in Brazil. Transbound. Emerg. Dis. 2018, 65, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Blomström, A.L.; Belák, S.; Fossum, C.; McKillen, J.; Allan, G.; Wallgren, P.; Berg, M. Detection of a novel porcine boca-like virus in the background of porcine circovirus type 2 induced postweaning multisystemic wasting syndrome. Virus Res. 2009, 146, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Smith, C.; Bishop, B.; Stewart, C.; Simonson, R. Complete genome sequence of a porcine polyomavirus from nasal swabs of pigs with respiratory disease. Genome Announc. 2018, 6, e00344-18. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Naoi, Y.; Kishimoto, M.; Masuda, T.; Tanabe, H.; Ito, M.; Niira, K.; Haga, K.; Asano, K.; Tsuchiaka, S.; et al. Identification of further diversity among posaviruses. Arch. Virol. 2016, 161, 3541–3548. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Collin, E.; Peddireddi, L.; Clement, T.; Gauger, P.; Hause, B.M. Genetic diversity in envelope genes of contemporary U.S. porcine reproductive and respiratory syndrome virus strains influences viral antigenicity. Res. Vet. Sci. 2017, 115, 432–441. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.; Xia, X.Q.; Chen, Q.; Bade, S.A.; Yoon, K.J.; Harmon, K.M.; Gauger, P.C.; Main, R.G.; Li, G. High-throughput whole genome sequencing of Porcine reproductive and respiratory syndrome virus from cell culture materials and clinical specimens using next-generation sequencing technology. J. Vet. Diagn. Investig. 2017, 29, 41–50. [Google Scholar] [CrossRef]
- Niira, K.; Ito, M.; Masuda, T.; Saitou, T.; Abe, T.; Komoto, S.; Sato, M.; Yamasato, H.; Kishimoto, M.; Naoi, Y.; et al. Whole genome sequences of Japanese porcine species C rotaviruses reveal a high diversity of genotypes of individual genes and will contribute to a comprehensive, generally accepted classification system. Infect. Genet. Evol. 2016, 44, 106–113. [Google Scholar] [CrossRef]
- Phan, T.G.; Giannitti, F.; Rossow, S.; Marthaler, D.; Knutson, T.; Li, L.; Deng, X.; Resende, T.; Vannucci, F.; Delwart, E. Detection of a novel circovirus PCV3 in pigs with cardiac and multi-systemic inflammation. Virol. J. 2016, 13, 184. [Google Scholar] [CrossRef]
- Alekseev, K.P.; Penin, A.A.; Mukhin, A.N.; Khametova, K.M.; Grebennikova, T.V.; Yuzhakov, A.G.; Moskvina, A.S.; Musienko, M.I.; Raev, S.A.; Mishin, A.M.; et al. Genome characterization of a pathogenic porcine rotavirus B strain identified in Buryat republic, Russia in 2015. Pathogens 2018, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Clilverd, H.; Cortey, M.; Martín-Valls, G.E.; Franzo, G.; Darwich, L.; Martín, M.; Mateu, E. Full-genome characterization by deep sequencing of rotavirus A isolates from outbreaks of neonatal diarrhoea in pigs in Spain. Vet. Microbiol. 2018, 227, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, W.; Cui, L.; Shen, Q.; Hua, X. Metagenomic identification, genetic characterization and genotyping of porcine sapoviruses. Infect. Genet. Evol. 2018, 62, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Masembe, C.; Michuki, G.; Onyango, M.; Rumberia, C.; Norling, M.; Bishop, R.P.; Djikeng, A.; Kemp, S.J.; Orth, A.; Skilton, R.A.; et al. Viral metagenomics demonstrates that domestic pigs are a potential reservoir for Ndumu virus. Virol. J. 2012, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Schlottau, K.; Schulze, C.; Bilk, S.; Hanke, D.; Höper, D.; Beer, M.; Hoffmann, B. Detection of a Novel Bovine Astrovirus in a Cow with Encephalitis. Transbound. Emerg. Dis. 2016, 63, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Baechlein, C.; Fischer, N.; Grundhoff, A.; Alawi, M.; Indenbirken, D.; Postel, A.; Baron, A.L.; Offinger, J.; Becker, K.; Beineke, A.; et al. Identification of a Novel Hepacivirus in Domestic Cattle from Germany. J. Virol. 2015, 89, 7007–7015. [Google Scholar] [CrossRef] [PubMed]
- Bauermann, F.V.; Joshi, L.R.; Mohr, K.A.; Kutish, G.F.; Meier, P.; Chase, C.; Christopher-Hennings, J.; Diel, D.G. A novel bovine papillomavirus type in the genus Dyokappapapillomavirus. Arch. Virol. 2017, 162, 3225–3228. [Google Scholar] [CrossRef]
- De Souza, W.M.; Dennis, T.; Fumagalli, M.J.; Araujo, J.; Sabino-Santos, G.; Maia, F.G.M.; Acrani, G.O.; Carrasco, A.D.O.T.; Romeiro, M.F.; Modha, S.; et al. Novel parvoviruses from wild and domestic animals in Brazil provide new insights into parvovirus distribution and diversity. Viruses 2018, 10, 143. [Google Scholar] [CrossRef]
- Nagai, M.; Omatsu, T.; Aoki, H.; Kaku, Y.; Belsham, G.J.; Haga, K.; Naoi, Y.; Sano, K.; Umetsu, M.; Shiokawa, M.; et al. Identification and complete genome analysis of a novel bovine picornavirus in Japan. Virus Res. 2015, 210, 205–212. [Google Scholar] [CrossRef]
- Hoffmann, B.; Scheuch, M.; Höper, D.; Jungblut, R.; Holsteg, M.; Schirrmeier, H.; Eschbaumer, M.; Goller, K.V.; Wernike, K.; Fischer, M.; et al. Novel orthobunyavirus in cattle, Europe, 2011. Emerg. Infect. Dis. 2012, 18, 469–472. [Google Scholar] [CrossRef]
- Masuda, T.; Nagai, M.; Yamasato, H.; Tsuchiaka, S.; Okazaki, S.; Katayama, Y.; Oba, M.; Nishiura, N.; Sassa, Y.; Omatsu, T.; et al. Identification of novel bovine group A rotavirus G15P[1 4] strain from epizootic diarrhea of adult cows by de novo sequencing using a next-generation sequencer. Vet. Microbiol. 2014, 171, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Pankovics, P.; Delwart, E.; Boros, Á. Identification of a novel astrovirus in domestic sheep in Hungary. Arch. Virol. 2012, 157, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, F.; Schlottau, K.; Scholes, S.; Courtenay, A.; Hoffmann, B.; Höper, D.; Beer, M. A novel astrovirus associated with encephalitis and ganglionitis in domestic sheep. Transbound. Emerg. Dis. 2017, 64, 677–682. [Google Scholar] [CrossRef]
- Chen, G.Q.; Zhuang, Q.Y.; Wang, K.C.; Liu, S.; Shao, J.Z.; Jiang, W.M.; Hou, G.Y.; Li, J.P.; Yu, J.M.; Li, Y.P.; et al. Identification and Survey of a Novel Avian Coronavirus in Ducks. PLoS ONE 2013, 8, e72918. [Google Scholar] [CrossRef]
- Boros, Á.; Polgár, B.; Pankovics, P.; Fenyvesi, H.; Engelmann, P.; Phan, T.G.; Delwart, E.; Reuter, G. Multiple divergent picobirnaviruses with functional prokaryotic Shine-Dalgarno ribosome binding sites present in cloacal sample of a diarrheic chicken. Virology 2018, 525, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Boros, Á.; Nemes, C.; Pankovics, P.; Kapusinszky, B.; Delwart, E.; Reuter, G. Identification and complete genome characterization of a novel picornavirus in Turkey (Meleagris gallopavo). J. Gen. Virol. 2012, 93, 2171–2182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boros, Á.; Nemes, C.; Pankovics, P.; Kapusinszky, B.; Delwart, E.; Reuter, G. Genetic characterization of a novel picornavirus in turkeys (Meleagris gallopavo) distinct from turkey galliviruses and megriviruses and distantly related to the members of the genus Avihepatovirus. J. Gen. Virol. 2013, 94, 1496–1509. [Google Scholar] [CrossRef][Green Version]
- Yu, J.M.; Li, J.S.; Ao, Y.Y.; Duan, Z.J. Detection of novel viruses in porcine fecal samples from China. Virol. J. 2013, 10, 39. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Hause, B.M. Detection and characterization of a novel genotype of porcine astrovirus 4 from nasal swabs from pigs with acute respiratory disease. Arch. Virol. 2016, 161, 2575–2579. [Google Scholar] [CrossRef]
- Yang, W.Z.; Yu, J.M.; Li, J.S.; Cheng, W.X.; Huang, C.P.; Duan, Z.J. Genome characterization of a novel porcine bocavirus. Arch. Virol. 2012, 157, 2125–2132. [Google Scholar] [CrossRef]
- Liu, L.; Schwarz, L.; Ullman, K.; Ahola, H.; Qiu, Y.; Ma, Z.; Hennig-Pauka, I. Identification of a novel bufavirus in domestic pigs by a viral metagenomic approach. J. Gen. Virol. 2016, 97, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Palinski, R.; Piñeyro, P.; Shang, P.; Yuan, F.; Guo, R.; Fang, Y.; Byers, E.; Hause, B.M. A Novel Porcine Circovirus Distantly Related to Known Circoviruses Is Associated with Porcine Dermatitis and Nephropathy Syndrome and Reproductive Failure. J. Virol. 2017, 91, e01879-16. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Katayama, Y.; Tsuchiaka, S.; Omatsu, T.; Murata, Y.; Ohya, K.; Makino, S.; Nagai, M.; Mizutani, T. Discovery of genome of an immunodeficiency-associated virus-like virus from pig feces in Japan. Jpn. J. Vet. Res. 2018, 66, 53–56. [Google Scholar]
- Schirtzinger, E.E.; Suddith, A.W.; Hause, B.M.; Hesse, R.A. First identification of porcine parvovirus 6 in North America by viral metagenomic sequencing of serum from pigs infected with porcine reproductive and respiratory syndrome virus. Virol. J. 2015, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Palinski, R.M.; Mitra, N.; Hause, B.M. Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes 2016, 52, 564–567. [Google Scholar] [CrossRef]
- Arruda, B.L.; Arruda, P.H.; Magstadt, D.R.; Schwartz, K.J.; Dohlman, T.; Schleining, J.A.; Patterson, A.R.; Visek, C.A.; Victoria, J.G. Identification of a divergent lineage porcine pestivirus in nursing piglets with congenital tremors and reproduction of disease following experimental inoculation. PLoS ONE 2016, 11, e0150104. [Google Scholar] [CrossRef]
- Naoi, Y.; Kishimoto, M.; Masuda, T.; Ito, M.; Tsuchiaka, S.; Sano, K.; Yamasato, H.; Omatsu, T.; Aoki, H.; Furuya, T.; et al. Characterization and phylogenetic analysis of a novel picornavirus from swine feces in Japan. Arch. Virol. 2016, 161, 1685–1690. [Google Scholar] [CrossRef]
- Hause, B.M.; Hesse, R.A.; Anderson, G.A. Identification of a novel Picornavirales virus distantly related to posavirus in swine feces. Virus Genes 2015, 51, 144–147. [Google Scholar] [CrossRef]
- Cotten, M.; Koopmans, M. Next-generation sequencing and norovirus. Future Virol. 2016, 11, 719–722. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Day, M.J.; Breitschwerdt, E.; Cleaveland, S.; Karkare, U.; Khanna, C.; Kirpensteijn, J.; Kuiken, T.; Lappin, M.R.; McQuiston, J.; Mumford, E.; et al. Surveillance of zoonotic infectious disease transmitted by small companion animals. Emerg. Infect. Dis. 2012, 18, e1. [Google Scholar] [CrossRef]
- Goede, D.; Morrison, R.B. Production impact & time to stability in sow herds infected with porcine epidemic diarrhea virus (PEDV). Prev. Vet. Med. 2016, 123, 202–207. [Google Scholar] [PubMed]
- Gallardo, M.C.; Reoyo, A.D.L.T.; Fernández-Pinero, J.; Iglesias, I.; Muñoz, M.J.; Arias, M.L. African swine fever: A global view of the current challenge. Porc. Health Manag. 2015, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- FAO. Food and Agricultural Organization of the United Nations Statistical Database. Available online: http://www.fao.org/faostat/en/#home (accessed on 15 November 2019).
- Ganter, M. Zoonotic risks from small ruminants. Vet. Microbiol. 2015, 181, 53–65. [Google Scholar] [CrossRef]
- Villabruna, N.; Koopmans, M.P.G.; de Graaf, M. Animals as reservoir for human norovirus. Viruses 2019, 11, 478. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Amimo, J.O.; Saif, L.J. Porcine rotaviruses: Epidemiology, immune responses and control strategies. Viruses 2017, 9, 48. [Google Scholar] [CrossRef]
- Simmonds, P. Methods for virus classification and the challenge of incorporating metagenomic sequence data. J. Gen. Virol. 2015, 96, 1193–1206. [Google Scholar] [CrossRef]
- Mande, S.S.; Mohammed, M.H.; Ghosh, T.S. Classification of metagenomic sequences: Methods and challenges. Brief. Bioinform. 2012, 13, 669–681. [Google Scholar] [CrossRef]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Aarestrup, F.M.; Koopmans, M.G. Sharing Data for Global Infectious Disease Surveillance and Outbreak Detection. Trends Microbiol. 2016, 24, 241–245. [Google Scholar] [CrossRef]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of virus metagenomic classification methods and their biological applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.E.J.; Brierley, L.; McCaffery, C.; Lycett, S. Assessing the epidemic potential of RNA and DNA viruses. Emerg. Infect. Dis. 2016, 22, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Seilmaier, M.; Corman, V.M.; Hartmann, W.; Scheible, G.; Sack, S.; Guggemos, W.; Kallies, R.; Muth, D.; Junglen, S.; et al. Clinical features and virological analysis of a case of Middle East respiratory syndrome coronavirus infection. Lancet Infect. Dis. 2013, 13, 745–751. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. Comment: The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed]
- Wielinga, P.R.; Hendriksen, R.S.; Aarestrup, F.M.; Lund, O.; Smits, S.L.; Koopmans, M.P.G.; Schlundt, J. Global Microbial Identifier. In Applied Genomics of Foodborne Pathogens; Springer: Berlin, Germany, 2017. [Google Scholar]
- Temmam, S.; Davoust, B.; Berenger, J.M.; Raoult, D.; Desnues, C. Viral metagenomics on animals as a tool for the detection of zoonoses prior to human infection? Int. J. Mol. Sci. 2014, 15, 10377–10397. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm Animal Type | Known Animal Viruses Found by mNGS Studies | References |
|---|---|---|
| Cattle | Bovine adenovirus | [44,48,49,50] |
| Bovine coronavirus | [44,50,51,52] | |
| Bovine papillomavirus | [53,54] | |
| Bovine parvovirus | [44,48,50,52,53,54,55,56] | |
| Bovine respiratory syncytial virus | [44] | |
| Bovine rhinitis A virus | [44,48,49] | |
| Bovine rhinitis B virus | [44,48,49,52,57] | |
| Bovine viral diarrhoea virus | [44,47] | |
| Enterovirus | [44,47,50,58] | |
| Hepatitis E virus | [47] | |
| Herpesvirus | [44,48,50,52,54,58] | |
| Influenza D virus | [48,59] | |
| Kobuvirus | [47] | |
| Norovirus | [58,60] | |
| Rotavirus | [47,58,61] | |
| Small ruminants | Enterovirus | [62] |
| Orf virus | [63,64] | |
| Rotavirus | [64,65] | |
| Poultry | Avastrovirus | [66,67,68,69,70] |
| Aveparvovirus | [69] | |
| Chicken anaemia virus | [68] | |
| Gallivirus | [70,71] | |
| Gammacoronavirus | [69,70] | |
| Influenza A virus | [25] | |
| Megrivirus | [69,70,71,72,73] | |
| Rotavirus | [67,70,72,74] | |
| Sicinivirus | [71,72] | |
| Tremovirus | [68,70] | |
| Pigs | African swine fever virus | [75] |
| Bocaparvovirus | [24,51,76,77,78,79,80,81] | |
| Enterovirus | [46,76,77,78,82,83,84,85,86,87,88,89,90,91,92,93] | |
| Hepatitis E virus | [45,46] | |
| Influenza A virus | [46] | |
| Kobuvirus | [46,76,77,78,79,80,84,87,88,91,92,94,95] | |
| Porcine adenovirus | [46,94] | |
| Porcine circovirus | [24,30,46,79,94,95,96,97,98] | |
| Porcine cytomegalovirus | [94,95,99] | |
| Porcine epidemic diarrhea virus | [46,79,84,88,92,100] | |
| Porcine reproductive and respiratory syndrome virus | [95,101,102] | |
| Porcine respiratory coronavirus | [46] | |
| Rotavirus | [24,80,82,83,84,87,90,92,100,103,104,105,106] | |
| Sapelovirus | [30,76,77,78,79,80,82,84,87,88,90,92,93,94] | |
| Sapovirus | [30,76,77,78,84,87,88,90,92,93,107] | |
| Torque teno virus | [24,30,46,79,94,95,108] |
| Farm Animal Type | Novel Viruses Found by mNGS Studies | References |
|---|---|---|
| Cattle | Astrovirus | [109] |
| CRESS-DNA virus | [50] | |
| Hepacivirus | [110] | |
| Nidovirus | [52] | |
| Papillomavirus | [111] | |
| Parvovirus | [112] | |
| Picornavirus | [113] | |
| Orthobunyavirus | [114] | |
| Rotavirus | [115] | |
| Small ruminants | Astrovirus | [116,117] |
| Poultry | Coronavirus | [118] |
| Picobirnavirus | [42,119] | |
| Picornavirus | [120,121] | |
| Pigs | Astrovirus | [122,123] |
| Bocavirus/Bocavirus-like | [98,122,124] | |
| Bufavirus | [125] | |
| Circovirus/Circovirus-like | [40,104,126] | |
| Enterovirus/Enterovirus-like | [85,89,92,93] | |
| Immunodeficiency-associated stool virus | [127] | |
| Ljungan-like viruses | [122] | |
| Parvovirus | [128,129] | |
| Porcine stool-associated circular virus | [82] | |
| Posavirus | [100] | |
| Pestivirus | [41,130] | |
| Picornavirus | [131] | |
| Picornavirales | [132] | |
| Teschovirus/Teschovirus-like | [90] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwok, K.T.T.; Nieuwenhuijse, D.F.; Phan, M.V.T.; Koopmans, M.P.G. Virus Metagenomics in Farm Animals: A Systematic Review. Viruses 2020, 12, 107. https://doi.org/10.3390/v12010107
Kwok KTT, Nieuwenhuijse DF, Phan MVT, Koopmans MPG. Virus Metagenomics in Farm Animals: A Systematic Review. Viruses. 2020; 12(1):107. https://doi.org/10.3390/v12010107
Chicago/Turabian StyleKwok, Kirsty T. T., David F. Nieuwenhuijse, My V. T. Phan, and Marion P. G. Koopmans. 2020. "Virus Metagenomics in Farm Animals: A Systematic Review" Viruses 12, no. 1: 107. https://doi.org/10.3390/v12010107
APA StyleKwok, K. T. T., Nieuwenhuijse, D. F., Phan, M. V. T., & Koopmans, M. P. G. (2020). Virus Metagenomics in Farm Animals: A Systematic Review. Viruses, 12(1), 107. https://doi.org/10.3390/v12010107