
Block-And-Lock: New Horizons for a Cure for HIV-1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
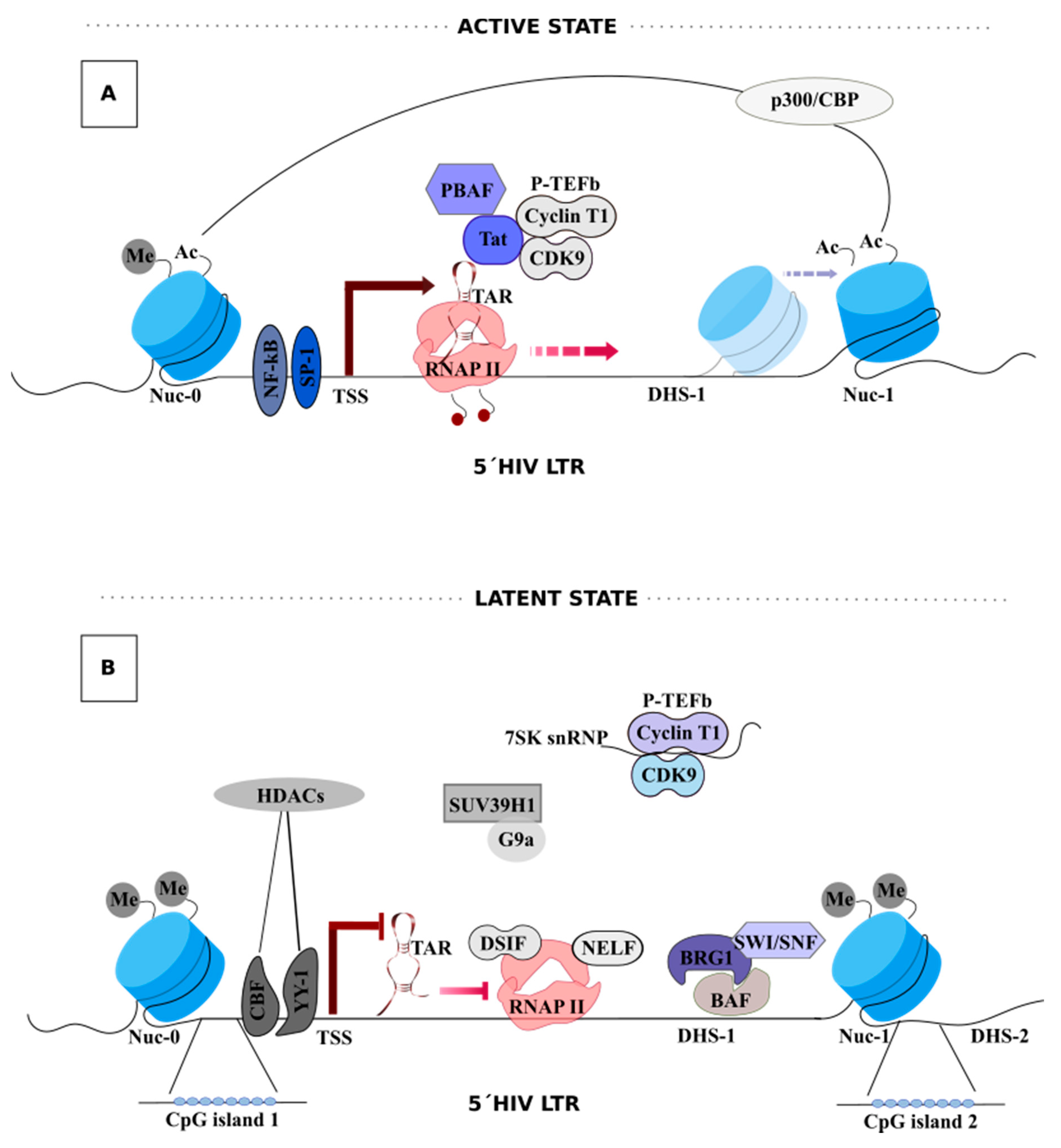
2. HIV-1 Latency
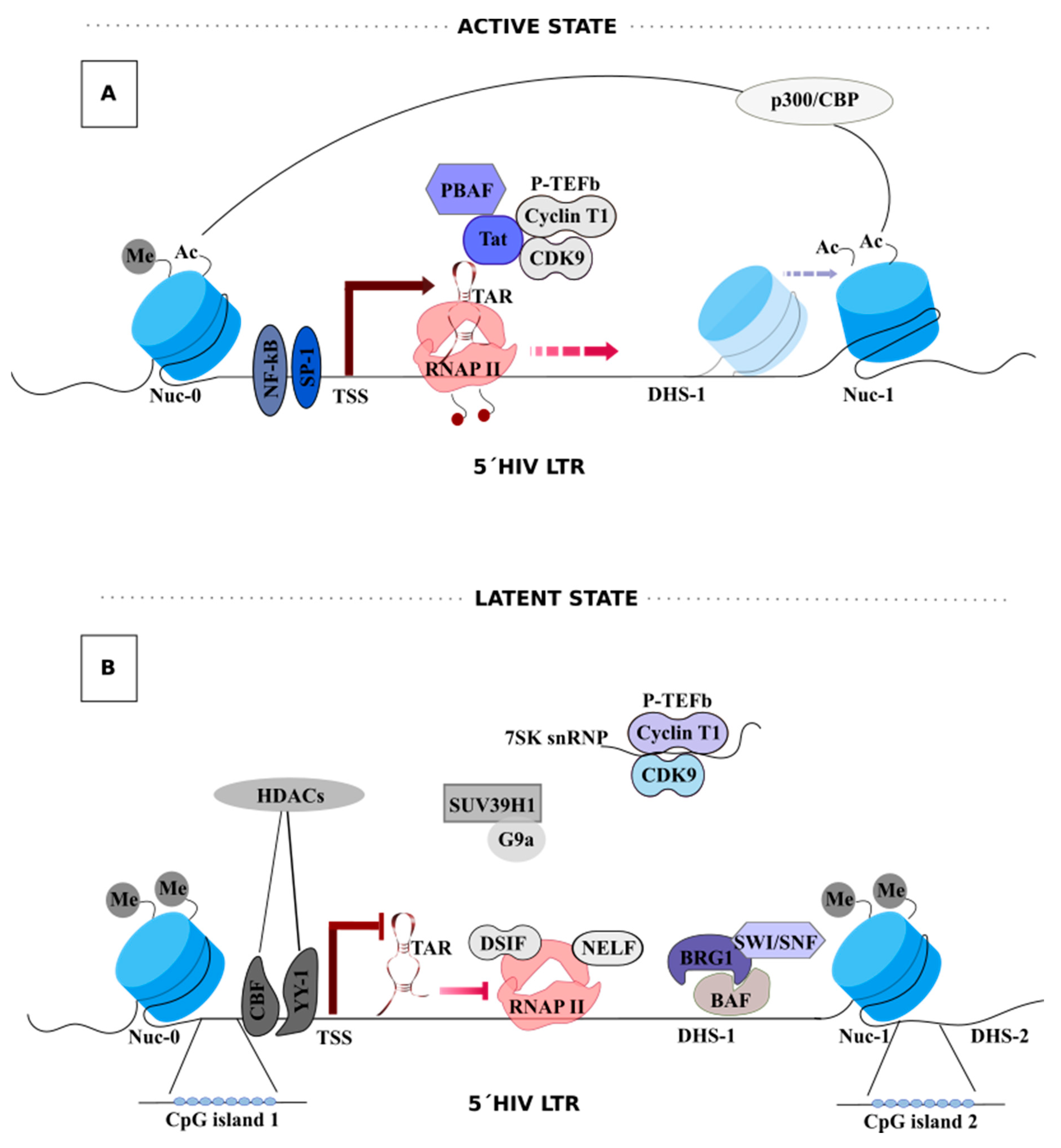
3. HIV-1 Transcription
Tat-Mediated HIV-1 Transcription
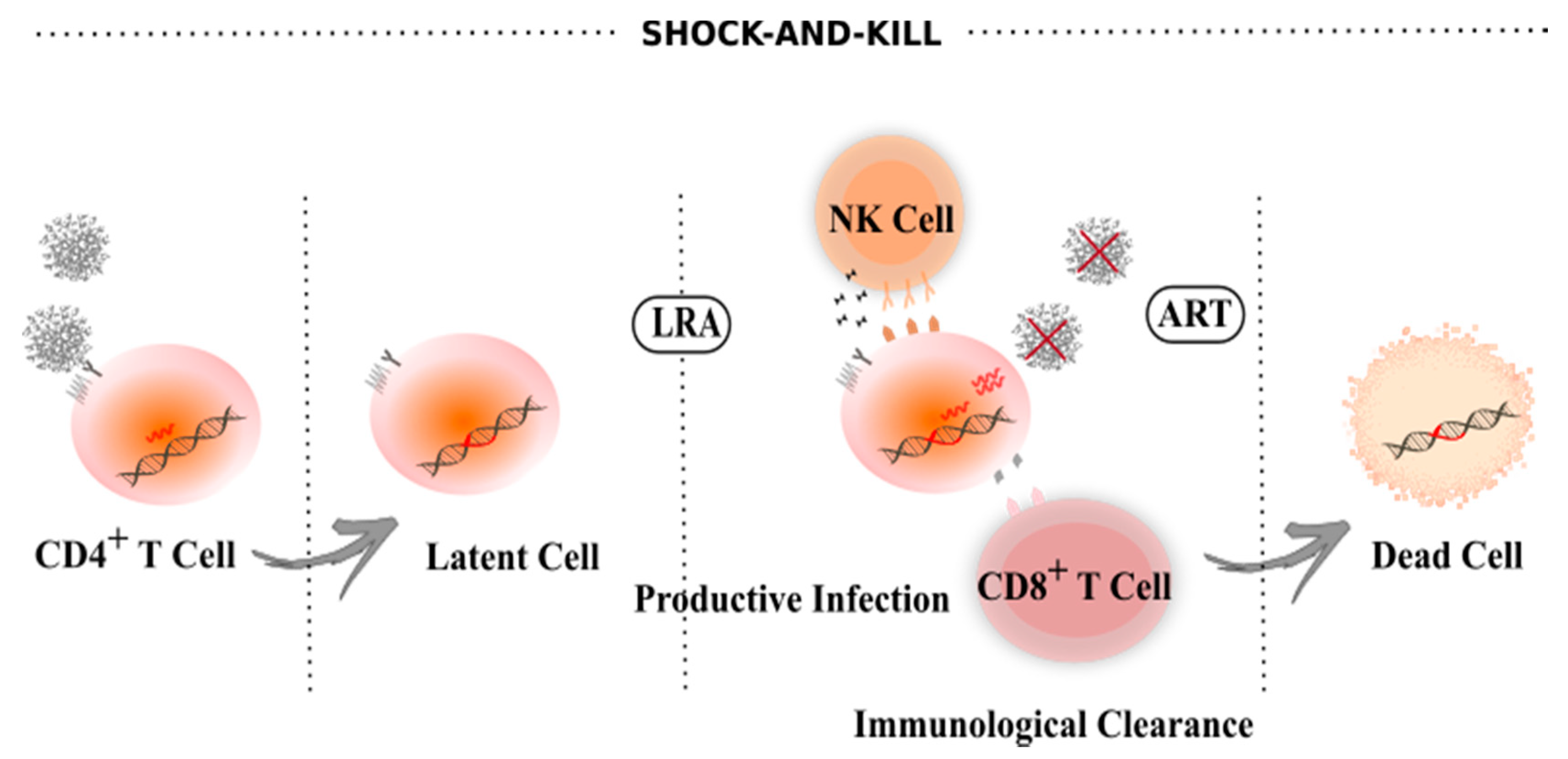
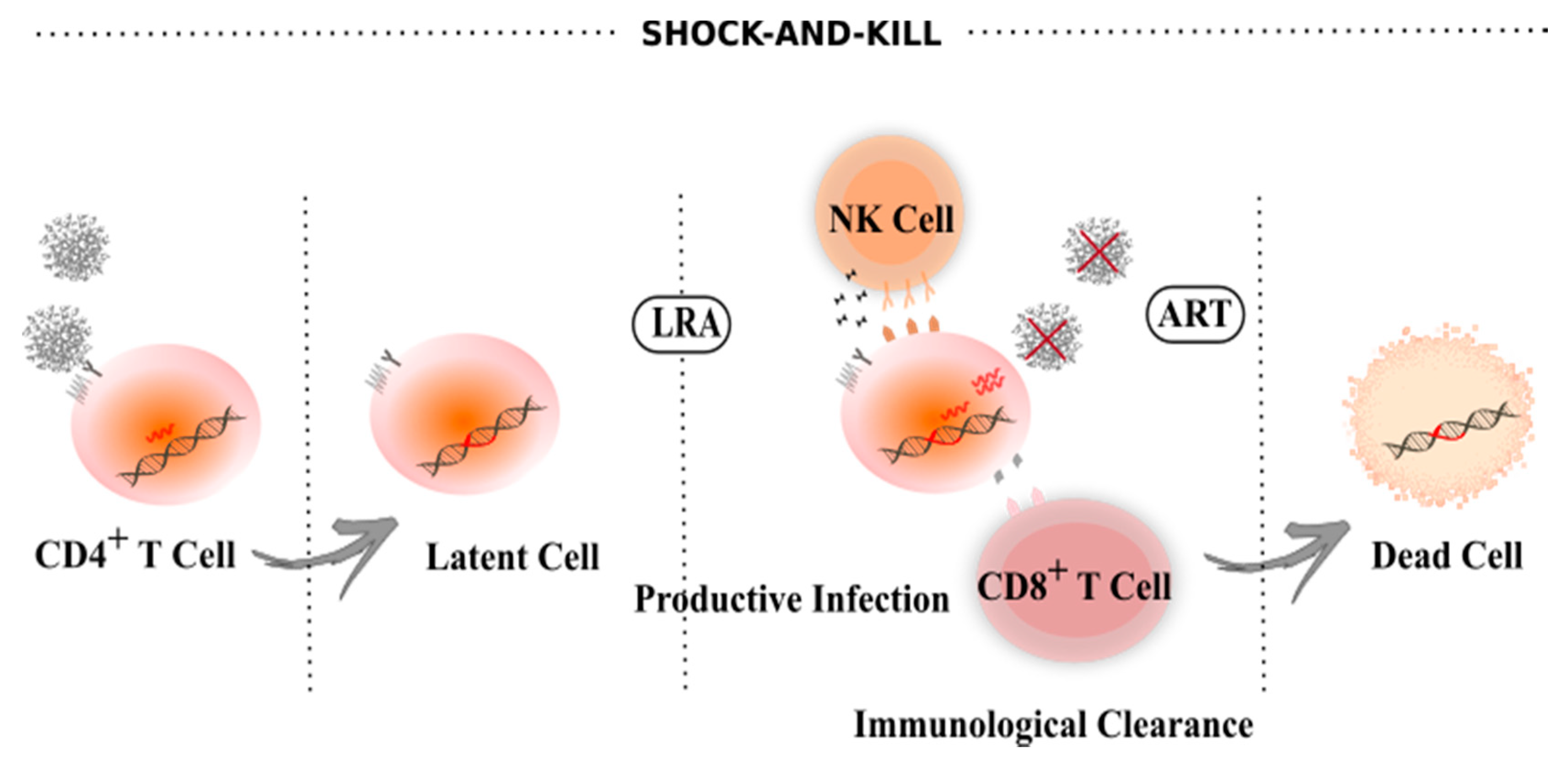
4. Latency Reversal Agents
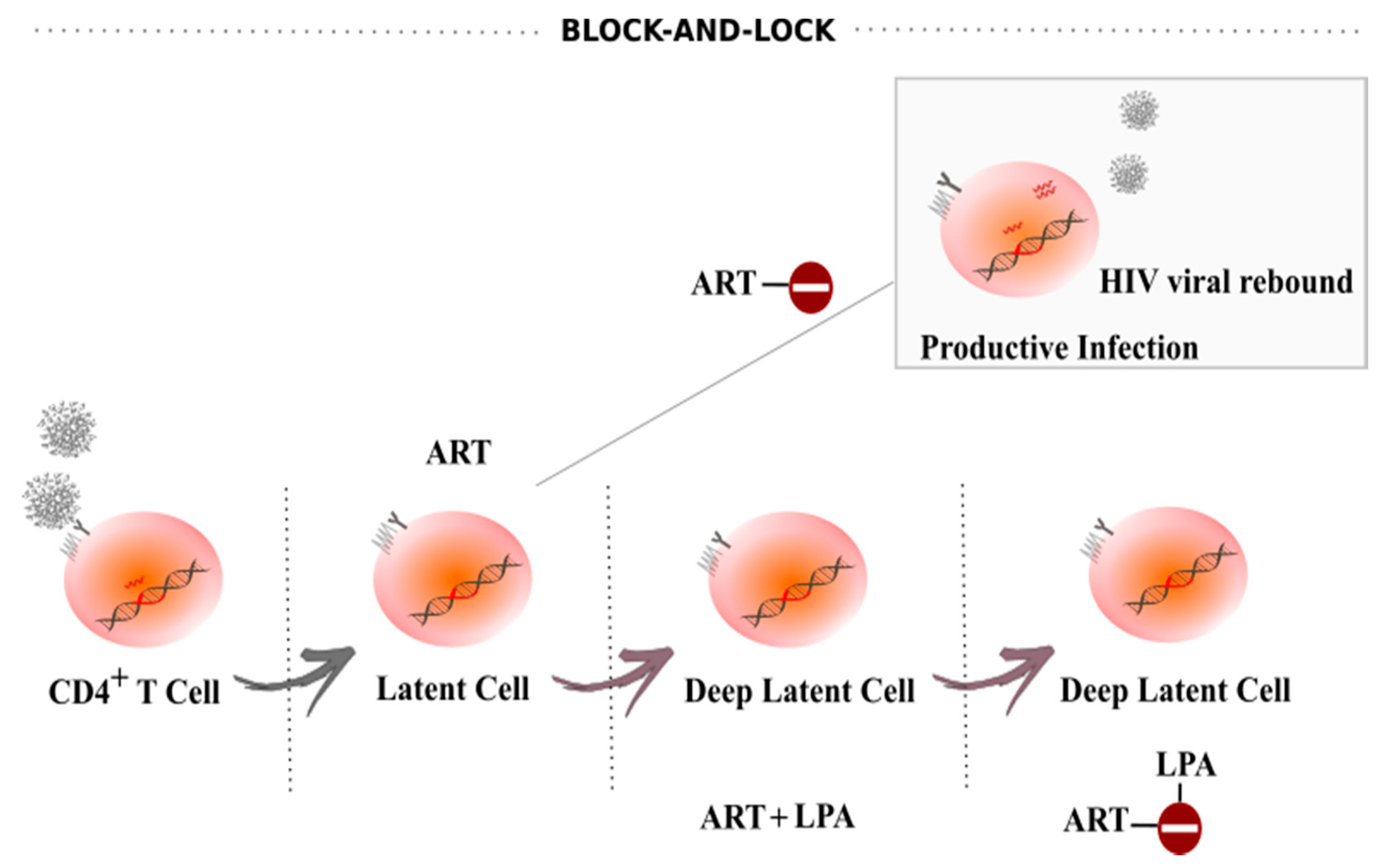
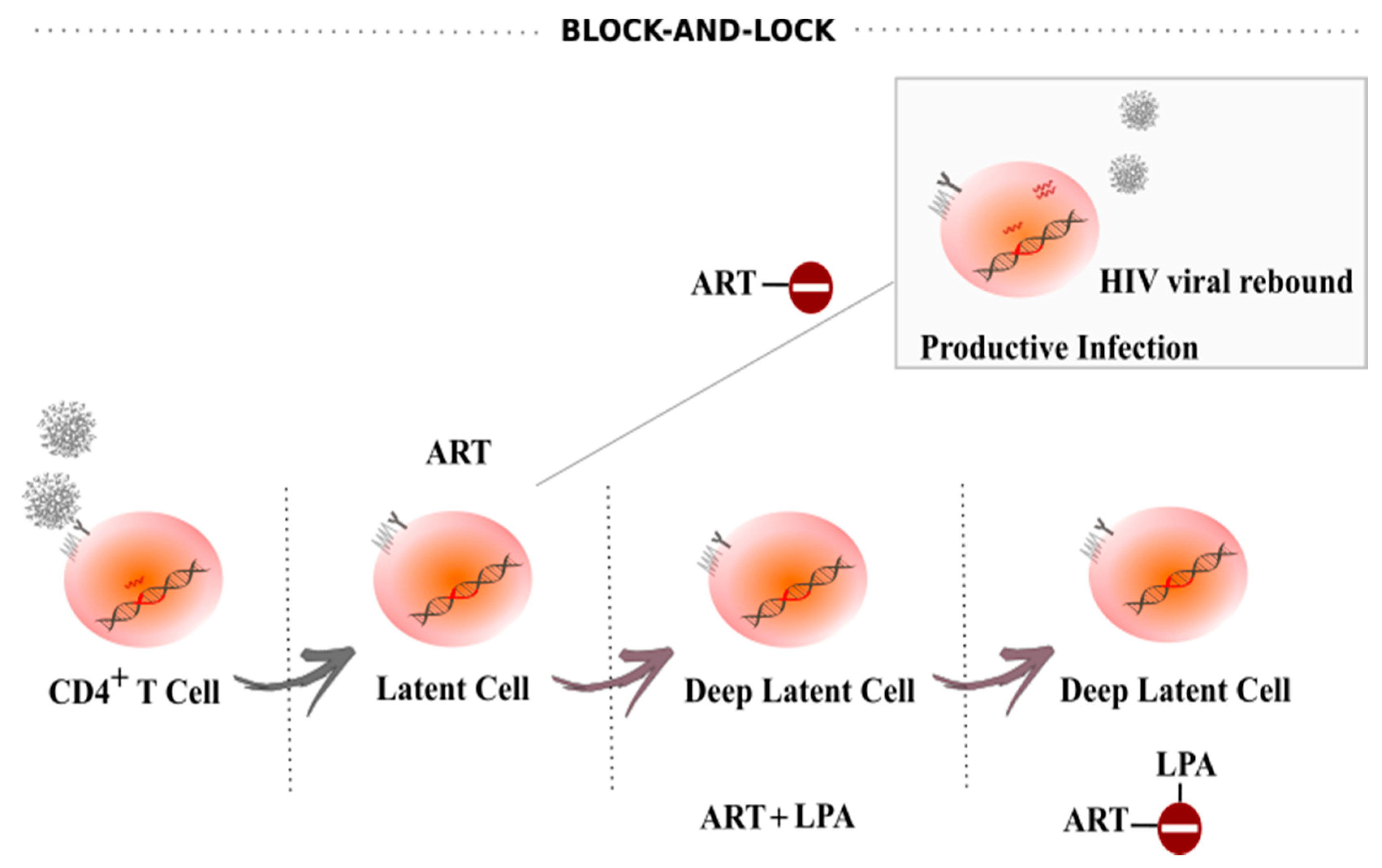
5. Block-And-Lock
5.1. Tat
5.2. Facilitates Chromatin Transcription (FACT) Complex
5.3. mTOR Complex (mTORC)
5.4. Bromodomain-Containing Protein 4 (BRD4)
5.5. Heat Shock Protein 90 (HSP90)
6. Other Sterilizing/Functional Cure Strategies
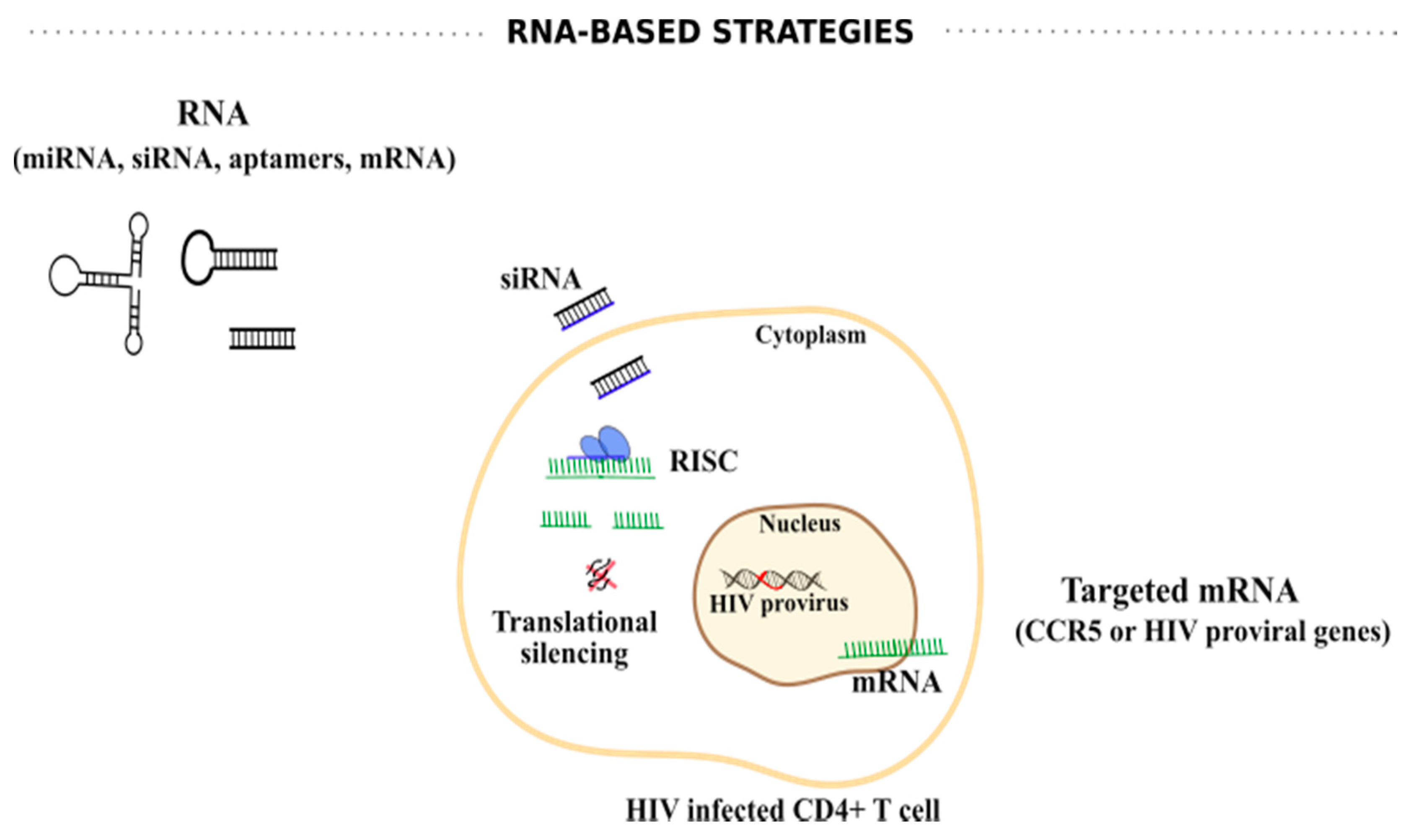
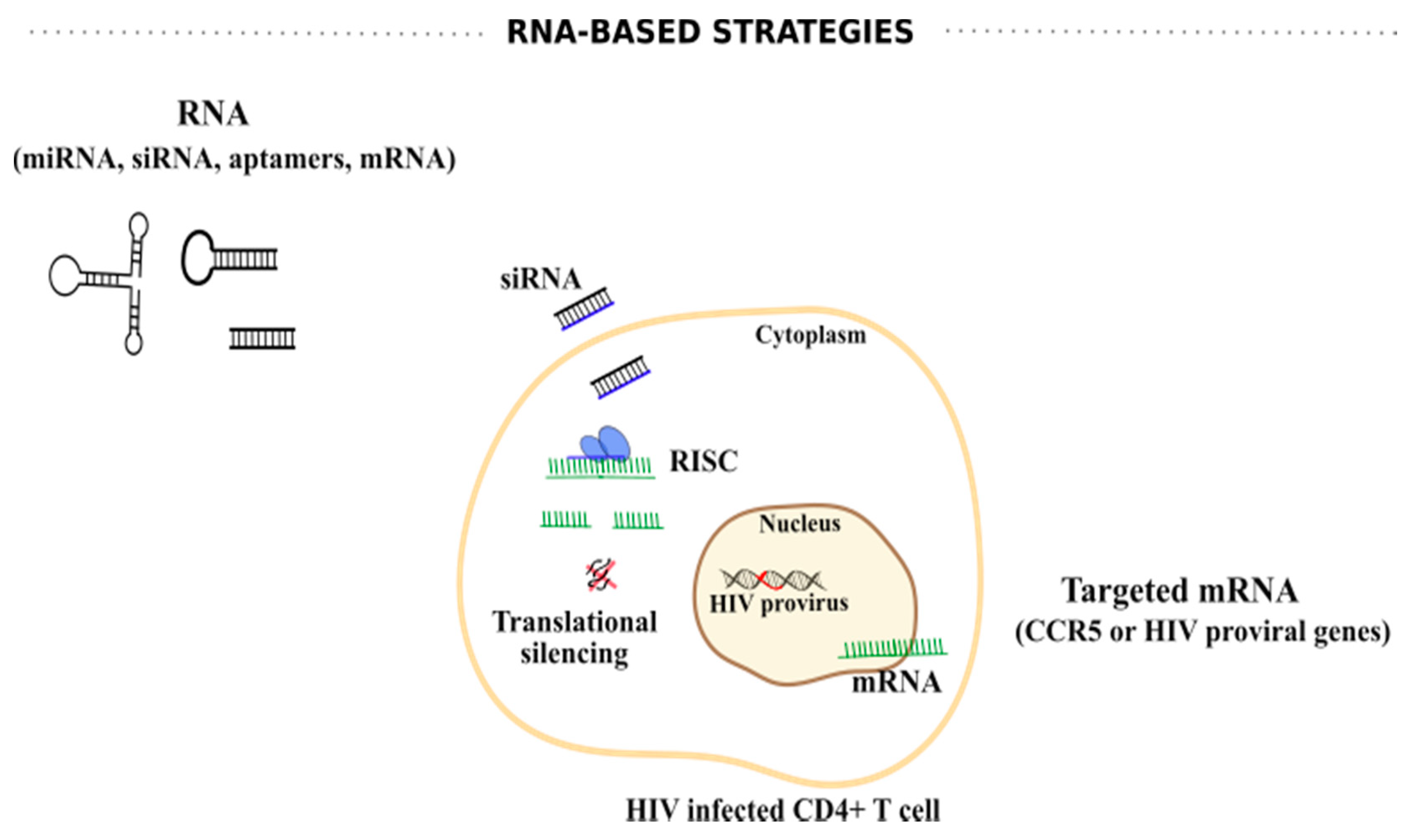
6.1. RNA-Based Strategies
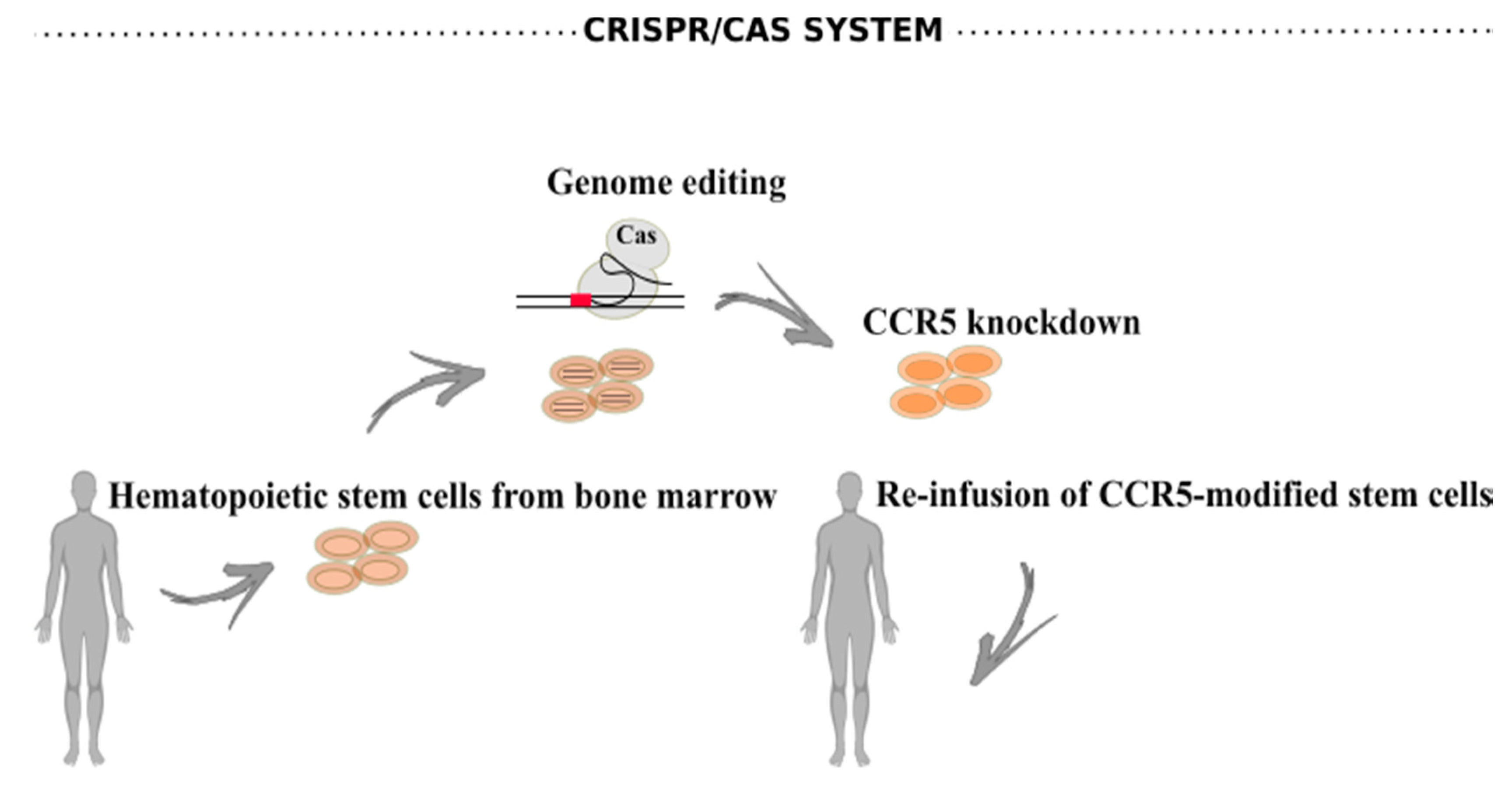
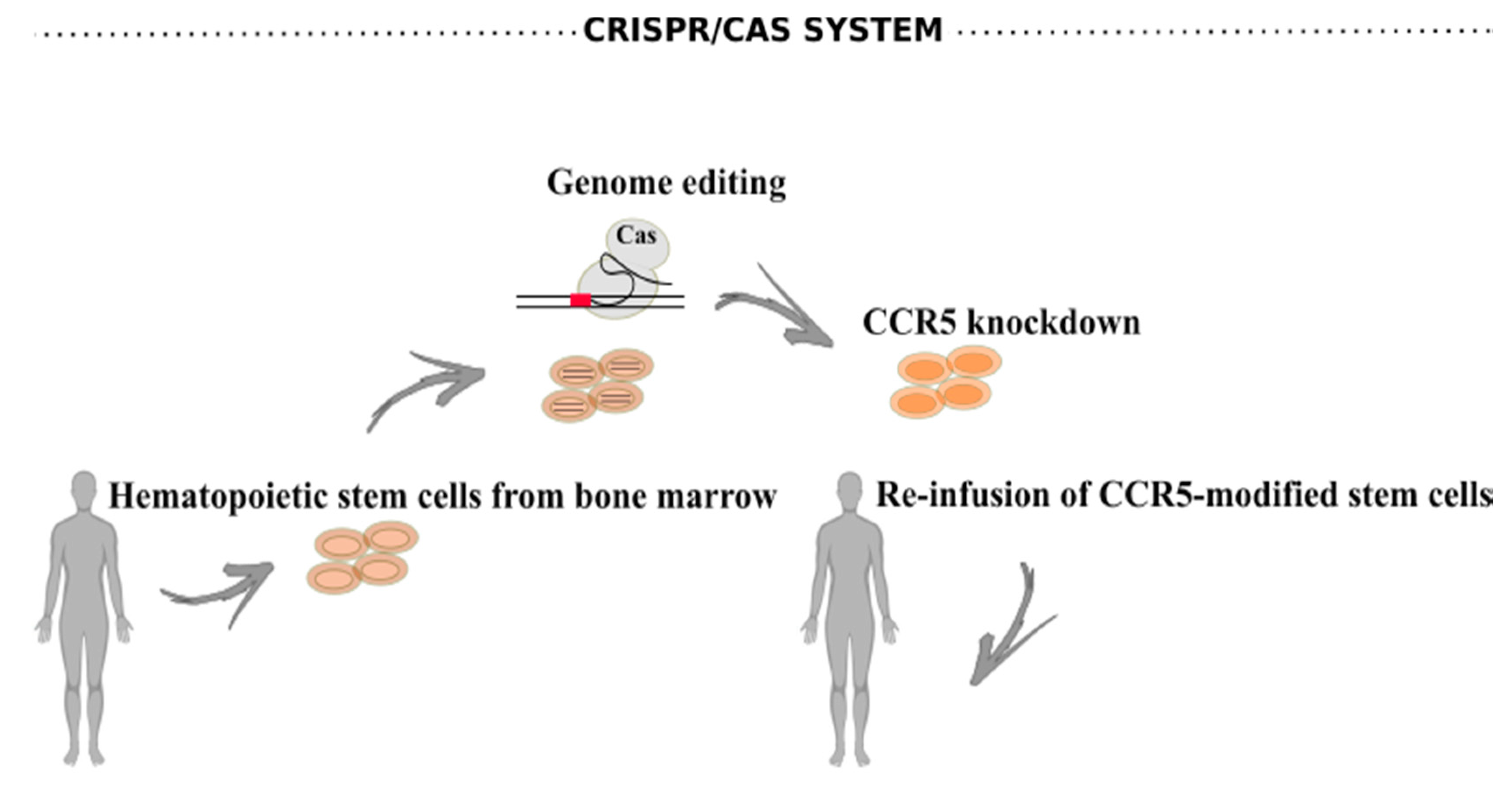
6.2. CRISPR/Cas Systems
7. Discussion
Funding
Conflicts of Interest
References
- World Health Organization (WHO). HIV-AIDS; Fact sheets; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Hamlyn, E.; Ewings, F.M.; Porter, K.; Cooper, D.A.; Tambussi, G.; Schechter, M.; Pedersen, C.; Okulicz, J.F.; McClure, M.; Babiker, A.; et al. Plasma HIV Viral Rebound following Protocol-Indicated Cessation of ART Commenced in Primary and Chronic HIV Infection. PLoS ONE 2012, 7, e43754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cihlar, T.; Fordyce, M. Current status and prospects of HIV treatment. Curr. Opin. Virol. 2016, 18, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrami, H.; Budoff, M.; Haberlen, S.A.; Rezaeian, P.; Ketlogetswe, K.; Tracy, R.; Palella, F.; Witt, M.D.; McConnell, M.V.; Kingsley, L.; et al. Inflammatory markers associated with subclinical coronary artery disease: The multicenter AIDS cohort study. J. Am. Heart Assoc. 2016, 5, e003371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Sun, B.; Gupta, A.; Rempel, H.; Pulliam, L. Monocyte exosomes induce adhesion molecules and cytokines via activation of NF-κB in endothelial cells. FASEB J. 2016, 30, 3097–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelesidis, T.; Kendall, M.A.; Yang, O.O.; Hodis, H.N.; Currier, J.S. Biomarkers of Microbial Translocation and Macrophage Activation: Association With Progression of Subclinical Atherosclerosis in HIV-1 Infection. J. Infect. Dis. 2012, 206, 1558–1567. [Google Scholar] [CrossRef]
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 drug resistance and resistance testing. Infect. Genet. Evol. 2016, 46, 292–307. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.I.; Castillo-Mancilla, J.R.; Tashima, K.T.; Landovitz, R.L. Advances in Long-Acting Agents for the Treatment of HIV Infection. Drugs 2020, 80, 535–545. [Google Scholar] [CrossRef]
- Meyers, K.; Rodriguez, K.; Moeller, R.W.; Gratch, I.; Markowitz, M.; Halkitis, P.N. High interest in a long-acting injectable formulation of pre-exposure prophylaxis for HIV in young men who have sex with men in NYC: A P18 cohort substudy. PLoS ONE 2014, 9, e114700. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, I.; Hashemi, F.B. Strategies to eradicate HIV from infected patients: Elimination of latent provirus reservoirs. Cell. Mol. Life Sci. 2019, 76, 3583–3600. [Google Scholar] [CrossRef] [Green Version]
- Stan, R.; Zaia, J.A. Practical considerations in gene therapy for HIV cure. Curr. HIV/AIDS Rep. 2014, 11, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müß, A.; Allers, K.; Schneider, T.; Hofmann, J.; Külcherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 delta32/delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Okulicz, J.F.; Lambotte, O. Epidemiology and clinical characteristics of elite controllers. Curr. Opin. HIV AIDS 2011, 6, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef]
- Cockerham, L.R.; Hatano, H.; Deeks, S.G. Post-Treatment Controllers: Role in HIV “Cure” Research. Curr. HIV/AIDS Rep. 2016, 13, 1–9. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV Latency: The Road to an HIV Cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Deng, K.; Gao, H.; Xing, S.; Capoferri, A.A.; Durand, C.M.; Rabi, S.A.; Laird, G.M.; Kim, M.; Hosmane, N.N.; et al. Transcriptional Reprogramming during Effector-to-Memory Transition Renders CD4+ T Cells Permissive for Latent HIV-1 Infection. Immunity 2017, 47, 766–775.e3. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, S.D.; Lu, H.K.; Chang, J.J.; Rhodes, A.; Lewin, S.R.; Cameron, P.U. The Pathway To Establishing HIV Latency Is Critical to How Latency Is Maintained and Reversed. J. Virol. 2018, 92, e02225-17. [Google Scholar] [CrossRef] [Green Version]
- Rong, L.; Perelson, A.S. Modeling HIV persistence, the latent reservoir, and viral blips. J. Theor. Biol. 2009, 260, 308–331. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.J.; Kwon, K.J.; Farber, D.L.; Siliciano, R.F. The Latent Reservoir for HIV-1: How Immunologic Memory and Clonal Expansion Contribute to HIV-1 Persistence. J. Immunol. 2016, 197, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananworanich, J.; Dubé, K.; Chomont, N. How does the timing of antiretroviral therapy initiation in acute infection affect HIV reservoirs? Curr. Opin. HIV AIDS 2015, 10, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, Z.M.; Kazer, S.W.; Nkosi, T.; Ogunshola, F.; Muema, D.M.; Anmole, G.; Swann, S.A.; Moodley, A.; Dong, K.; Reddy, T.; et al. Augmentation of HIV-specific T cell function by immediate treatment of hyperacute HIV-1 infection. Sci. Transl. Med. 2019, 11, eaau0528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540. [Google Scholar] [CrossRef] [Green Version]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Massanella, M.; Richman, D.D. Measuring the latent reservoir in vivo. J. Clin. Investig. 2016, 126, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative Analysis of Measures of Viral Reservoirs in HIV-1 Eradication Studies. PLoS Pathog. 2013, 9, e1003174. [Google Scholar] [CrossRef]
- Symons, J.; Cameron, P.U.; Lewin, S.R. HIV integration sites and implications for maintenance of the reservoir. Curr. Opin. HIV AIDS 2018, 13, 152–159. [Google Scholar] [CrossRef]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antivir. Res. 2019, 166, 19–34. [Google Scholar] [CrossRef]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Castro-Gonzalez, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for HIV Cure: The Latent Reservoir. AIDS Res. Hum. Retrovir. 2018, 34, 739–759. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Ussel, M.; Vandergeeten, C.; Marchionni, L.; Chomont, N.; Romerio, F. High Levels of CD2 Expression Identify HIV-1 Latently Infected Resting Memory CD4+ T Cells in Virally Suppressed Subjects. J. Virol. 2013, 87, 9148–9158. [Google Scholar] [CrossRef] [Green Version]
- Darcis, G.; Kootstra, N.A.; Hooibrink, B.; van Montfort, T.; Maurer, I.; Groen, K.; Jurriaans, S.; Bakker, M.; van Lint, C.; Berkhout, B.; et al. CD32+CD4+ T Cells Are Highly Enriched for HIV DNA and Can Support Transcriptional Latency. Cell Rep. 2020, 30, 2284–2296.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massanella, M.; Fromentin, R.; Chomont, N. Residual inflammation and viral reservoirs: Alliance against an HIV cure. Curr. Opin. HIV AIDS 2016, 11, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Simonetti, F.R.; Ho, Y.C. The forces driving clonal expansion of the HIV-1 latent reservoir. Virol. J. 2020, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Ne, E.; Palstra, R.J.; Mahmoudi, T. Transcription: Insights From the HIV-1 Promoter. Int. Rev. Cell Mol. Biol. 2018, 335, 191–243. [Google Scholar]
- Olson, A.; Basukala, B.; Wong, W.W.; Henderson, A.J. Targeting HIV-1 proviral transcription. Curr. Opin. Virol. 2019, 38, 89–96. [Google Scholar] [CrossRef]
- Kamori, D.; Ueno, T. HIV-1 tat and viral latency: What we can learn from naturally occurring sequence variations. Front. Microbiol. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodroski, J.; Rosen, C.; Wong-Staal, F.; Salahuddin, S.; Popovic, M.; Arya, S.; Gallo, R.; Haseltine, W. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science 1985, 227, 171–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, S.-Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Berkhout, B.; Silverman, R.H.; Jeang, K.-T. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 1989, 59, 273–282. [Google Scholar] [CrossRef]
- Bannwarth, S.; Gatignol, A. HIV-1 TAR RNA: The Target of Molecular Interactions Between the Virus and its Host. Curr. HIV Res. 2005, 3, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.-H.; Rana, T.M. DSIF and NELF Interact with RNA Polymerase II Elongation Complex and HIV-1 Tat Stimulates P-TEFb-mediated Phosphorylation of RNA Polymerase II and DSIF during Transcription Elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef] [Green Version]
- Price, D.H. P-TEFb, a Cyclin-Dependent Kinase Controlling Elongation by RNA Polymerase II. Mol. Cell. Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Barboric, M.; Nissen, R.M.; Kanazawa, S.; Jabrane-Ferrat, N.; Peterlin, B.M. NF-κB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 2001, 8, 327–337. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Moon, K.J.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- O’Brien, S.K.; Cao, H.; Nathans, R.; Ali, A.; Rana, T.M. P-TEFb Kinase Complex Phosphorylates Histone H1 to Regulate Expression of Cellular and HIV-1 Genes. J. Biol. Chem. 2010, 285, 29713–29720. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, M.; Chen, L.-F.; Chen, R. P-TEFb: Finding its ways to release promoter-proximally paused RNA polymerase II. Transcription 2018, 9, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. Tackling tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Garber, M.E.; Fang, S.-M.; Fischer, W.H.; Jones, K.A. A Novel CDK9-Associated C-Type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Brady, J.; Kashanchi, F. Tat gets the “Green” light on transcription initiation. Retrovirology 2005, 2, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdin, E.; Paras, P.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef]
- Verdin, E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 1991, 65, 6790–6799. [Google Scholar] [CrossRef] [Green Version]
- El Kharroubi, A.; Verdin, E. Protein-DNA interactions within DNase I-hypersensitive sites located downstream of the HIV-1 promoter. J. Biol. Chem. 1994, 269, 19916–19924. [Google Scholar]
- Tréand, C.; Du Chéné, I.; Brès, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar] [CrossRef]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Easley, R.; Carpio, L.; Dannenberg, L.; Choi, S.; Alani, D.; Van Duyne, R.; Guendel, I.; Klase, Z.; Agbottah, E.; Kehn-Hall, K.; et al. Transcription through the HIV-1 nucleosomes: Effects of the PBAF complex in Tat activated transcription. Virology 2010, 405, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusic, M.; Giacca, M. Regulation of HIV-1 Latency by Chromatin Structure and Nuclear Architecture. J. Mol. Biol. 2015, 427, 688–694. [Google Scholar] [CrossRef]
- He, G.; Margolis, D.M. Counterregulation of Chromatin Deacetylation and Histone Deacetylase Occupancy at the Integrated Promoter of Human Immunodeficiency Virus Type 1 (HIV-1) by the HIV-1 Repressor YY1 and HIV-1 Activator Tat. Mol. Cell. Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef] [PubMed]
- Coull, J.J.; Romerio, F.; Sun, J.-M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The Human Factors YY1 and LSF Repress the Human Immunodeficiency Virus Type 1 Long Terminal Repeat via Recruitment of Histone Deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef] [Green Version]
- Du Chéné, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [Green Version]
- Bednarik, D.P.; Cook, J.A.; Pitha, P.M. Inactivation of the HIV LTR by DNA CpG methylation: Evidence for a role in latency. EMBO J. 1990, 9, 1157–1164. [Google Scholar] [CrossRef]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef] [Green Version]
- Chávez, L.; Kauder, S.; Verdin, E. In vivo, in vitro, and in silico analysis of methylation of the HIV-1 provirus. Methods 2011, 53, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2′-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.K.; Moir, S.; Chun, T.-W.; Fauci, A.S. Paucity of HIV DNA Methylation in Latently Infected, Resting CD4+ T Cells from Infected Individuals Receiving Antiretroviral Therapy. J. Virol. 2012, 86, 5390–5392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, J.A.; Perez-Pinar, T.; Toro, C.; Sanz-Minguela, B.; Moreno, V.; Valencia, E.; Gomez-Hernando, C.; Rodes, B. Long-Term Nonprogressor and Elite Controller Patients Who Control Viremia Have a Higher Percentage of Methylation in Their HIV-1 Proviral Promoters than Aviremic Patients Receiving Highly Active Antiretroviral Therapy. J. Virol. 2012, 86, 13081–13084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Huang, K.; Jung, K.-J.; Cho, W.-K.; Klase, Z.; Kashanchi, F.; Pise-Masison, C.A.; Brady, J.N. Bromodomain Protein Brd4 Regulates Human Immunodeficiency Virus Transcription through Phosphorylation of CDK9 at Threonine 29. J. Virol. 2009, 83, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol. Cell 2017, 67, 1001–1012.e6. [Google Scholar] [CrossRef] [Green Version]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 Tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Schnölzer, M.; Garnica, J.; Fischle, W.; Emiliani, S.; Rackwitz, H.-R.; Verdin, E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999, 9, 1489–1493. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; de la Fuente, C.; Fu, P.; Wang, L.; Donnelly, R.; Wade, J.D.; Lambert, P.; Li, H.; Lee, C.-G.; Kashanchi, F. Acetylation of HIV-1 Tat by CBP/P300 Increases Transcription of Integrated HIV-1 Genome and Enhances Binding to Core Histones. Virology 2000, 277, 278–295. [Google Scholar] [CrossRef] [Green Version]
- Delannoy, A.; Poirier, M.; Bell, B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses 2019, 11, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, M.J.; Fiches, G.; Hayashi, T.; Zhu, J. Current Strategies for Elimination of HIV-1 Latent Reservoirs Using Chemical Compounds Targeting Host and Viral Factors. AIDS Res. Hum. Retrovir. 2019, 35, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Siliciano, R.F. Progress Toward HIV Eradication: Case Reports, Current Efforts, and the Challenges Associated with Cure. Annu. Rev. Med. 2016, 67, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Hashemi, P.; Sadowski, I. Diversity of small molecule HIV-1 latency reversing agents identified in low- and high-throughput small molecule screens. Med. Res. Rev. 2020, 40, 881–908. [Google Scholar] [CrossRef] [Green Version]
- Dash, P.K.; Kevadiya, B.D.; Su, H.; Banoub, M.G.; Gendelman, H.E. Pathways towards human immunodeficiency virus elimination. EBioMedicine 2020, 53, 102667. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV Transcription with Short-Course Vorinostat in HIV-Infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [Green Version]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.M.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef] [Green Version]
- Spivak, A.M.; Planelles, V. HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLOS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Dandekar, S. Targeting NF-κB Signaling with Protein Kinase C Agonists As an Emerging Strategy for Combating HIV Latency. AIDS Res. Hum. Retrovir. 2015, 31, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, A.M.; Bosque, A.; Balch, A.H.; Smyth, D.; Martins, L.; Planelles, V. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4 + T Cells from Aviremic Patients. Antimicrob. Agents Chemother. 2015, 59, 5984–5991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.K.; Thompson, G.R.; et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLOS Pathog. 2015, 11, e1005066. [Google Scholar] [CrossRef] [Green Version]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.S.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Hashemi, P.; Barreto, K.; Bernhard, W.; Lomness, A.; Honson, N.; Pfeifer, T.A.; Harrigan, P.R.; Sadowski, I. Compounds producing an effective combinatorial regimen for disruption of HIV-1 latency. EMBO Mol. Med. 2018, 10, 160–174. [Google Scholar] [CrossRef]
- Clutton, G.T.; Jones, R.B. Diverse impacts of HIV latency-reversing agents on CD8+ T-cell function: Implications for HIV cure. Front. Immunol. 2018, 9, 1452. [Google Scholar] [CrossRef]
- Leth, S.; Schleimann, M.H.; Nissen, S.K.; Højen, J.F.; Olesen, R.; Graversen, M.E.; Jørgensen, S.; Kjær, A.S.; Denton, P.W.; Mørk, A.; et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): A single-arm, phase 1B/2A trial. Lancet HIV 2016, 3, e463–e472. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, A.; Blanch-Lombarte, O.; Jimenez-Moyano, E.; Ouchi, D.; Mothe, B.; Peña, R.; Galvez, C.; Genescà, M.; Martinez-Picado, J.; Goulder, P.; et al. Antigen Production After Latency Reversal and Expression of Inhibitory Receptors in CD8+ T Cells Limit the Killing of HIV-1 Reactivated Cells. Front. Immunol. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Grau-Expósito, J.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badía, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLOS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Søgaard, O.S. Reversal of Latency as Part of a Cure for HIV-1. Trends Microbiol. 2016, 24, 90–97. [Google Scholar] [CrossRef]
- Gama, L.; Abreu, C.M.; Shirk, E.N.; Price, S.L.; Li, M.; Laird, G.M.; Pate, K.A.M.; Wietgrefe, S.W.; O’Connor, S.L.; Pianowski, L.; et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 2017, 31, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Pardons, M.; Fromentin, R.; Pagliuzza, A.; Routy, J.-P.; Chomont, N. Latency-Reversing Agents Induce Differential Responses in Distinct Memory CD4 T Cell Subsets in Individuals on Antiretroviral Therapy. Cell Rep. 2019, 29, 2783–2795.e5. [Google Scholar] [CrossRef]
- Liu, C.; Ma, X.; Liu, B.; Chen, C.; Zhang, H. HIV-1 functional cure: Will the dream come true? BMC Med. 2015, 13, 284. [Google Scholar] [CrossRef] [Green Version]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef]
- Rands, C.M.; Meader, S.; Ponting, C.P.; Lunter, G. 8.2% of the Human Genome Is Constrained: Variation in Rates of Turnover across Functional Element Classes in the Human Lineage. PLoS Genet. 2014, 10, e1004525. [Google Scholar] [CrossRef] [Green Version]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.-I.; Kubota, S.; Siomi, H.; Adachi, A.; Oroszlan, S.; Maki, M.; Hatanaka, M. A region of basic amino-acid cluster in HIV-1 Tat protein is essential forTrans-acting activity and nucleolar localization. Virus Genes 1989, 3, 99–110. [Google Scholar] [CrossRef]
- Razooky, B.S.; Pai, A.; Aull, K.; Rouzine, I.M.; Weinberger, L.S. A Hardwired HIV Latency Program. Cell 2015, 160, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, S.; Palu, G. Inhibitors of HIV-1 Tat-Mediated Transactivation. Curr. Med. Chem. 2006, 13, 1305–1315. [Google Scholar] [CrossRef]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An Analog of the Natural Steroidal Alkaloid Cortistatin A Potently Suppresses Tat-Dependent HIV Transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mediouni, S.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Clementz, M.; Kovach, M.D.; Mousseau, G.; De Vera, I.M.S.; Li, C.; Kojetin, D.J.; et al. The Tat inhibitor didehydro-cortistatin A suppresses SIV replication and reactivation. FASEB J. 2019, 33, 8280–8293. [Google Scholar] [CrossRef]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. MBio 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Jablonski, J.; Paris, J.; Clementz, M.; Thenin-Houssier, S.; McLaughlin, J.; Valente, S. Didehydro-Cortistatin A Inhibits HIV-1 Tat Mediated Neuroinflammation and Prevents Potentiation of Cocaine Reward in Tat Transgenic Mice. Curr. HIV Res. 2015, 13, 64–79. [Google Scholar] [CrossRef] [Green Version]
- Maragos, W.F.; Young, K.L.; Turchan, J.T.; Guseva, M.; Pauly, J.R.; Nath, A.; Cass, W.A. Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J. Neurochem. 2002, 83, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Dai, S.-M. A Chinese herb Tripterygium wilfordii Hook F in the treatment of rheumatoid arthritis: Mechanism, efficacy, and safety. Rheumatol. Int. 2011, 31, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Chen, X. Triptolide inhibits human immunodeficiency virus type 1 replication by promoting proteasomal degradation of Tat protein. Retrovirology 2014, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, J.; He, L.; Yu, Q. Triptolide (TPL) Inhibits Global Transcription by Inducing Proteasome-Dependent Degradation of RNA Polymerase II (Pol II). PLoS ONE 2011, 6, e23993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titov, D.V.; Gilman, B.; He, Q.-L.; Bhat, S.; Low, W.-K.; Dang, Y.; Smeaton, M.; Demain, A.L.; Miller, P.S.; Kugel, J.F.; et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 2011, 7, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Jean, M.; Huang, H.; Simpson, S.; Santoso, N.G.; Zhu, J. Screening of an FDA-approved compound library identifies levosimendan as a novel anti-HIV-1 agent that inhibits viral transcription. Antivir. Res. 2017, 146, 76–85. [Google Scholar] [CrossRef]
- Kasikcioglu, H.A.; Cam, N. A review of levosimendan in the treatment of heart failure. Vasc. Health Risk Manag. 2006, 2, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Alekseev, S.; Ayadi, M.; Brino, L.; Egly, J.-M.; Larsen, A.K.; Coin, F. A Small Molecule Screen Identifies an Inhibitor of DNA Repair Inducing the Degradation of TFIIH and the Chemosensitization of Tumor Cells to Platinum. Chem. Biol. 2014, 21, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, B.; Morel, M.; Margottin-Goguet, F.; Ramirez, B.C. Specific Inhibition of HIV Infection by the Action of Spironolactone in T Cells. J. Virol. 2016, 90, 10972–10980. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Santoso, N.; Power, D.; Simpson, S.; Dieringer, M.; Miao, H.; Gurova, K.; Giam, C.-Z.; Elledge, S.J.; Zhu, J. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J. Biol. Chem. 2015, 290, 27297–27310. [Google Scholar] [CrossRef] [Green Version]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer Compounds That Simultaneously Suppress NF- B and Activate p53 by Targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef]
- Jean, M.J.; Hayashi, T.; Huang, H.; Brennan, J.; Simpson, S.; Purmal, A.; Gurova, K.; Keefer, M.C.; Kobie, J.J.; Santoso, N.G.; et al. Curaxin CBL0100 Blocks HIV-1 Replication and Reactivation through Inhibition of Viral Transcriptional Elongation. Front. Microbiol. 2017, 8, 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Jiang, Y. Key factors in mTOR regulation. Cell. Mol. Life Sci. 2010, 67, 239–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, R.J.O.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Amoroso, A.; Davis, C.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV -chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [Green Version]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Chan, J.K.; Greene, W.C. Dynamic roles for NF-κB in HTLV-I and HIV-1 retroviral pathogenesis. Immunol. Rev. 2012, 246, 286–310. [Google Scholar] [CrossRef]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Liao, Q.; Chen, J.; Zhang, L.; He, Q.; Zhu, H.; Zhang, X.; Xu, J. TSC1 and DEPDC5 regulate HIV-1 latency through the mTOR signaling pathway. Emerg. Microbes Infect. 2018, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Invest. 2019, 129, 3361–3373. [Google Scholar] [CrossRef]
- Vozzolo, L.; Loh, B.; Gane, P.J.; Tribak, M.; Zhou, L.; Anderson, I.; Nyakatura, E.; Jenner, R.G.; Selwood, D.; Fassati, A. Gyrase B Inhibitor Impairs HIV-1 Replication by Targeting Hsp90 and the Capsid Protein. J. Biol. Chem. 2010, 285, 39314–39328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Stoddart, C.A. Impaired infectivity of ritonavir-resistant HIV is rescued by heat shock protein 90AB1. J. Biol. Chem. 2011, 286, 24581–24592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Choi, M.-S.; Inn, K.-S.; Kim, B.-J. Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a HSP90-dependent manner. Sci. Rep. 2016, 6, 28896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, I.; Low, J.S.; Weston, S.; Weinberger, M.; Zhyvoloup, A.; Labokha, A.A.; Corazza, G.; Kitson, R.A.; Moody, C.J.; Marcello, A.; et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl. Acad. Sci. USA 2014, 111, E1528–E1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Maidji, E.; Stoddart, C.A. Inhibition of Heat Shock Protein 90 Prevents HIV Rebound. J. Biol. Chem. 2016, 291, 10332–10346. [Google Scholar] [CrossRef] [Green Version]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-A.; Kim, J.; Sim, J.; Kim, S.-G.; Kook, Y.-H.; Park, C.-G.; Kim, H.-R.; Kim, B.-J. A telomerase-derived peptide regulates reactive oxygen species and hepatitis C virus RNA replication in HCV-infected cells via heat shock protein 90. Biochem. Biophys. Res. Commun. 2016, 471, 156–162. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Sung, T.-L.; Rice, A.P. miR-198 Inhibits HIV-1 Gene Expression and Replication in Monocytes and Its Mechanism of Action Appears To Involve Repression of Cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef] [Green Version]
- Chiang, K.; Sung, T.-L.; Rice, A.P. Regulation of Cyclin T1 and HIV-1 Replication by MicroRNAs in Resting CD4+ T Lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Feng, H.; Da, Q.; Jiang, H.; Chen, L.; Xie, L.; Huang, Q.; Xiong, H.; Luo, F.; Kang, L.; et al. Expression of HIV-encoded microRNA-TAR and its inhibitory effect on viral replication in human primary macrophages. Arch. Virol. 2016, 161, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, Q.; Binder, G.K.; Chen, Z.; Slepushkina, T.; Rossi, J.; Dropulic, B. Antisense-Mediated Inhibition of Human Immunodeficiency Virus (HIV) Replication by Use of an HIV Type 1-Based Vector Results in Severely Attenuated Mutants Incapable of Developing Resistance. J. Virol. 2004, 78, 7079–7088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Held, D.M. HIV-1 inactivation by nucleic acid aptamers. Front. Biosci. 2006, 11, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, K.-T.; Zhou, J.; Rossi, J. Aptamer-Based Therapeutics: New Approaches to Combat Human Viral Diseases. Pharmaceuticals 2013, 6, 1507–1542. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, H.; Li, S.; Zaia, J.; Rossi, J.J. Novel Dual Inhibitory Function Aptamer–siRNA Delivery System for HIV-1 Therapy. Mol. Ther. 2008, 16, 1481–1489. [Google Scholar] [CrossRef]
- Duclair, S.; Gautam, A.; Ellington, A.; Prasad, V.R. High-affinity RNA Aptamers Against the HIV-1 Protease Inhibit Both In Vitro Protease Activity and Late Events of Viral Replication. Mol. Ther. Nucleic Acids 2015, 4, e228. [Google Scholar] [CrossRef]
- Zhou, J.; Satheesan, S.; Li, H.; Weinberg, M.S.; Morris, K.V.; Burnett, J.C.; Rossi, J.J. Cell-Specific RNA Aptamer against Human CCR5 Specifically Targets HIV-1 Susceptible Cells and Inhibits HIV-1 Infectivity. Chem. Biol. 2015, 22, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Burnett, J.C.; Rossi, J.J. Aptamer–siRNA Chimeras for HIV. Adv. Exp. Med. Biol. 2015, 848, 211–234. [Google Scholar]
- Lee, N.S.; Dohjima, T.; Bauer, G.; Li, H.; Li, M.-J.; Ehsani, A.; Salvaterra, P.; Rossi, J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 2002, 20, 500–505. [Google Scholar] [CrossRef]
- Coburn, G.A.; Cullen, B.R. Potent and Specific Inhibition of Human Immunodeficiency Virus Type 1 Replication by RNA Interference. J. Virol. 2002, 76, 9225–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novina, C.D.; Murray, M.F.; Dykxhoorn, D.M.; Beresford, P.J.; Riess, J.; Lee, S.-K.; Collman, R.G.; Lieberman, J.; Shankar, P.; Sharp, P.A. siRNA-directed inhibition of HIV-1 infection. Nat. Med. 2002, 8, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, P.; de Vries, W.; Haasnoot, J.; ter Brake, O.; de Haan, P.; Berkhout, B. Inhibition of human immunodeficiency virus type 1 by RNA interference using long-hairpin RNA. Gene Ther. 2006, 13, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Jacque, J.M.; Triques, K.; Stevenson, M. Modulation of HIV-1 replication by RNA interference. Nature 2002, 418, 435–438. [Google Scholar] [CrossRef]
- Westerhout, E.M. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005, 33, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human Immunodeficiency Virus Type 1 Escapes from RNA Interference-Mediated Inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Nohtomi, K.; Onogi, T.; Uenishi, R.; Ui-Tei, K.; Saigo, K.; Takebe, Y. Optimal design and validation of antiviral siRNA for targeting HIV-1. Retrovirology 2007, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Ringpis, G.-E.E.; Shimizu, S.; Arokium, H.; Camba-Colón, J.; Carroll, M.V.; Cortado, R.; Xie, Y.; Kim, P.Y.; Sahakyan, A.; Lowe, E.L.; et al. Engineering HIV-1-Resistant T-Cells from Short-Hairpin RNA-Expressing Hematopoietic Stem/Progenitor Cells in Humanized BLT Mice. PLoS ONE 2012, 7, e53492. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, D.; Ramírez-Solís, R.; Garza-Elizondo, M.; Garza-Rodríguez, M.; Barrera-Saldana, H. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int. J. Mol. Med. 2019, 43, 1559–1574. [Google Scholar]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, P.; Uguccioni, M.; Bordoli, L.; Baggiolini, M.; Moser, B.; Chizzolini, C.; Dayer, J.-M. CCR5 is characteristic of Th1 lymphocytes. Nature 1998, 391, 344–345. [Google Scholar] [CrossRef] [PubMed]
- Ashmore-Harris, C.; Fruhwirth, G.O. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin. Transl. Med. 2020, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Zhang, T.; Li, F.; Yang, F.; Putatunda, R.; Young, W.-B.; Khalili, K.; Hu, W.; Zhang, Y. Functional screening of guide RNAs targeting the regulatory and structural HIV-1 viral genome for a cure of AIDS. AIDS 2016, 30, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef]
- Kordelas, L.; Verheyen, J.; Esser, S. Shift of HIV Tropism in Stem-Cell Transplantation with CCR5 Delta32 Mutation. N. Engl. J. Med. 2014, 371, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.L.; Parent, B.; Shen, M.; Plunkett, C. No time to waste—The ethical challenges created by CRISPR. EMBO Rep. 2015, 16, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M. CRISPR/Cas9 genome editing—New and old ethical issues arising from a revolutionary technology. Nanoethics 2016, 10, 139–159. [Google Scholar] [CrossRef]
- Normile, D. Shock greets claim of CRISPR-edited babies. Science 2018, 362, 978–979. [Google Scholar] [CrossRef]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune activation and HIV persistence: Implications for curative approaches to HIV infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [Green Version]
- Rouzine, I.M.; Weinberger, A.D.; Weinberger, L.S. An Evolutionary Role for HIV Latency in Enhancing Viral Transmission. Cell 2015, 160, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Marchi, E.; Kanapin, A.; Magiorkinis, G.; Belshaw, R. Unfixed Endogenous Retroviral Insertions in the Human Population. J. Virol. 2014, 88, 9529–9537. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, REVIEWS1017. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moranguinho, I.; Valente, S.T. Block-And-Lock: New Horizons for a Cure for HIV-1. Viruses 2020, 12, 1443. https://doi.org/10.3390/v12121443
Moranguinho I, Valente ST. Block-And-Lock: New Horizons for a Cure for HIV-1. Viruses. 2020; 12(12):1443. https://doi.org/10.3390/v12121443
Chicago/Turabian StyleMoranguinho, Ines, and Susana T. Valente. 2020. "Block-And-Lock: New Horizons for a Cure for HIV-1" Viruses 12, no. 12: 1443. https://doi.org/10.3390/v12121443
APA StyleMoranguinho, I., & Valente, S. T. (2020). Block-And-Lock: New Horizons for a Cure for HIV-1. Viruses, 12(12), 1443. https://doi.org/10.3390/v12121443