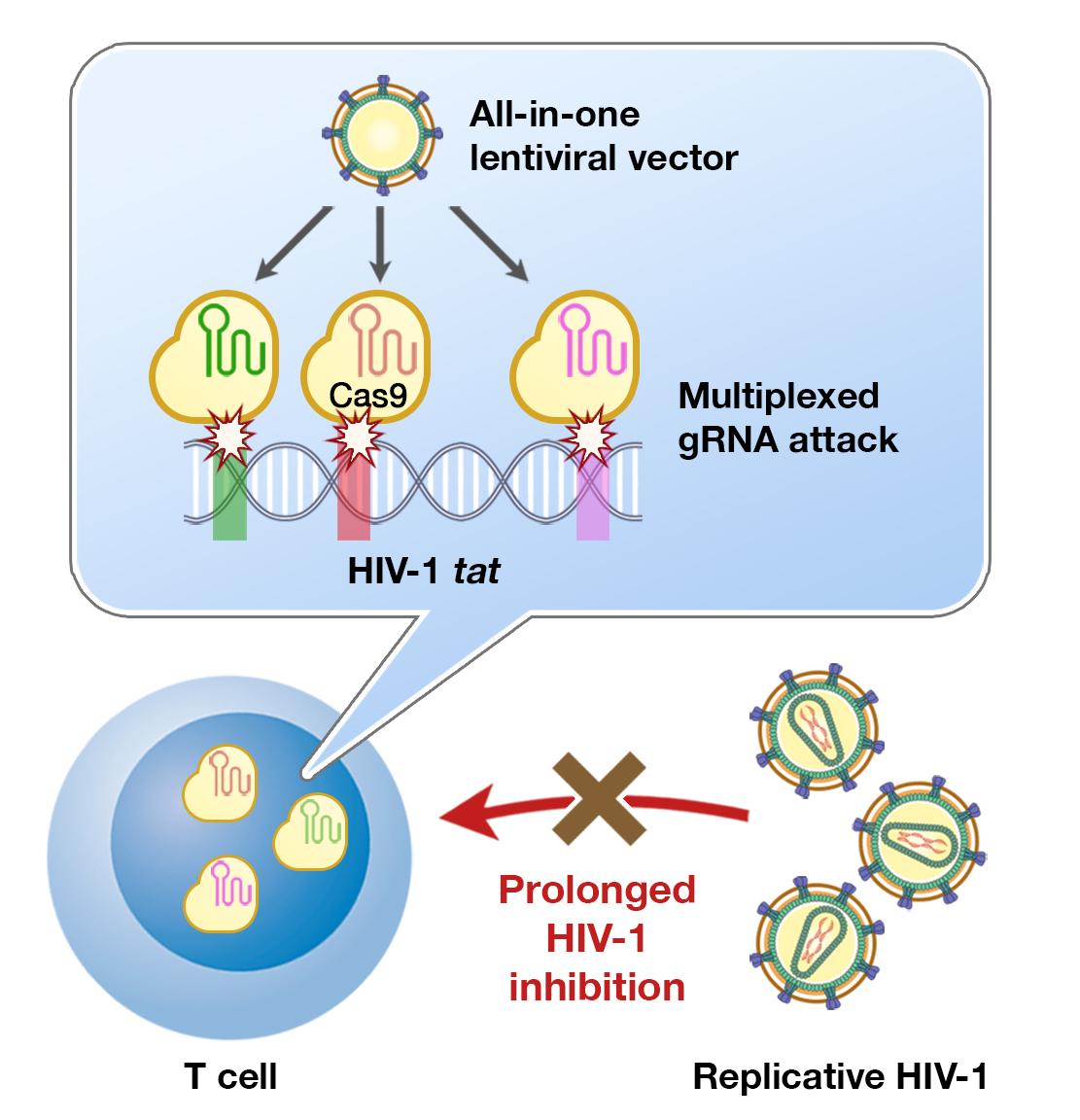
Multiplexed tat-Targeting CRISPR-Cas9 Protects T Cells from Acute HIV-1 Infection with Inhibition of Viral Escape



Abstract

1. Introduction
2. Materials and Methods
2.1. gRNA Designs and Plasmids
2.2. Cell Culture, Transfection and Transduction
2.3. HIV-1 Production and Infection
2.4. Western Blotting
2.5. On-Target Analyses
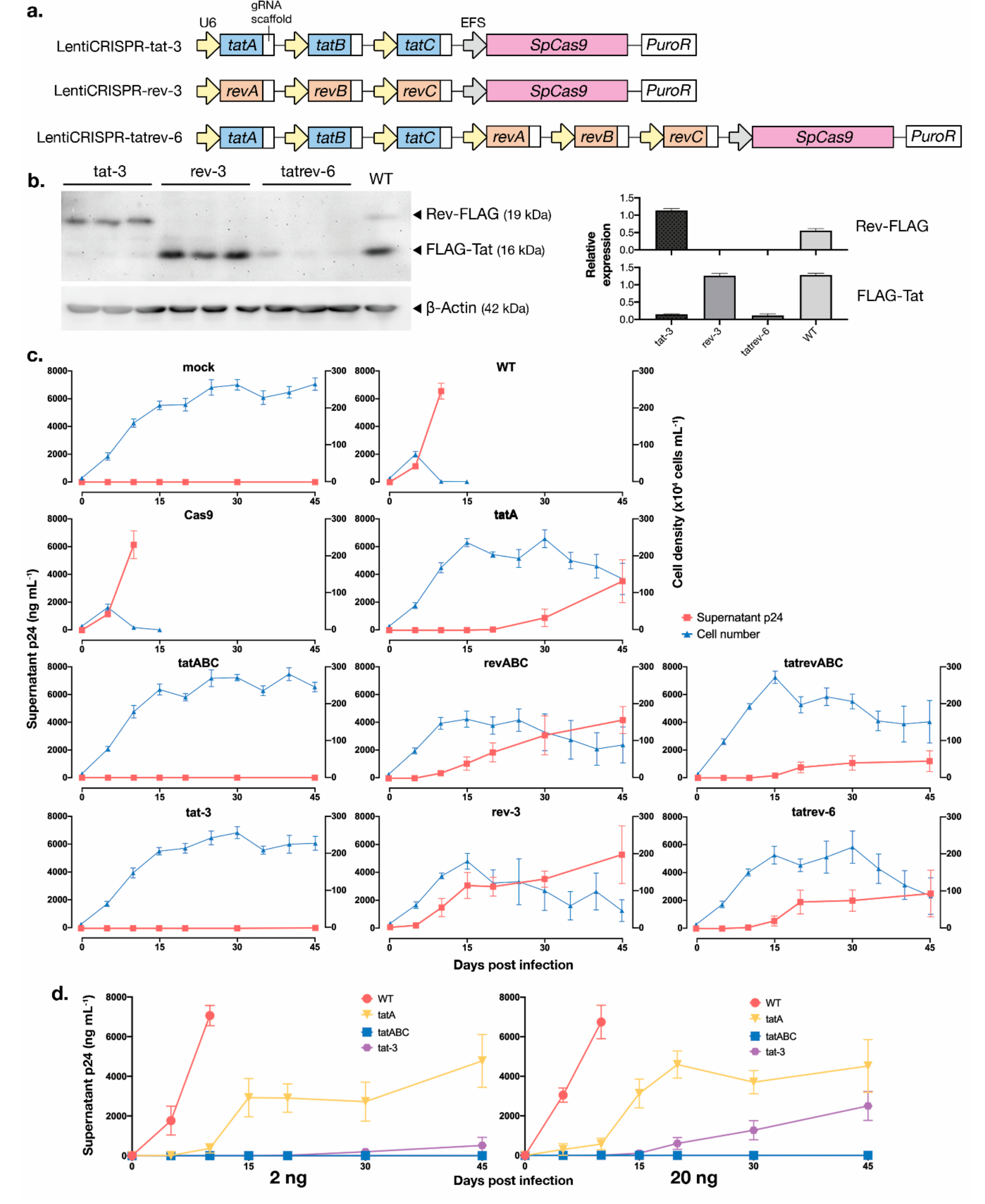
2.6. Development of Multiplexed, All-in-One CRISPR-Cas9 Lentiviral Vector
3. Results
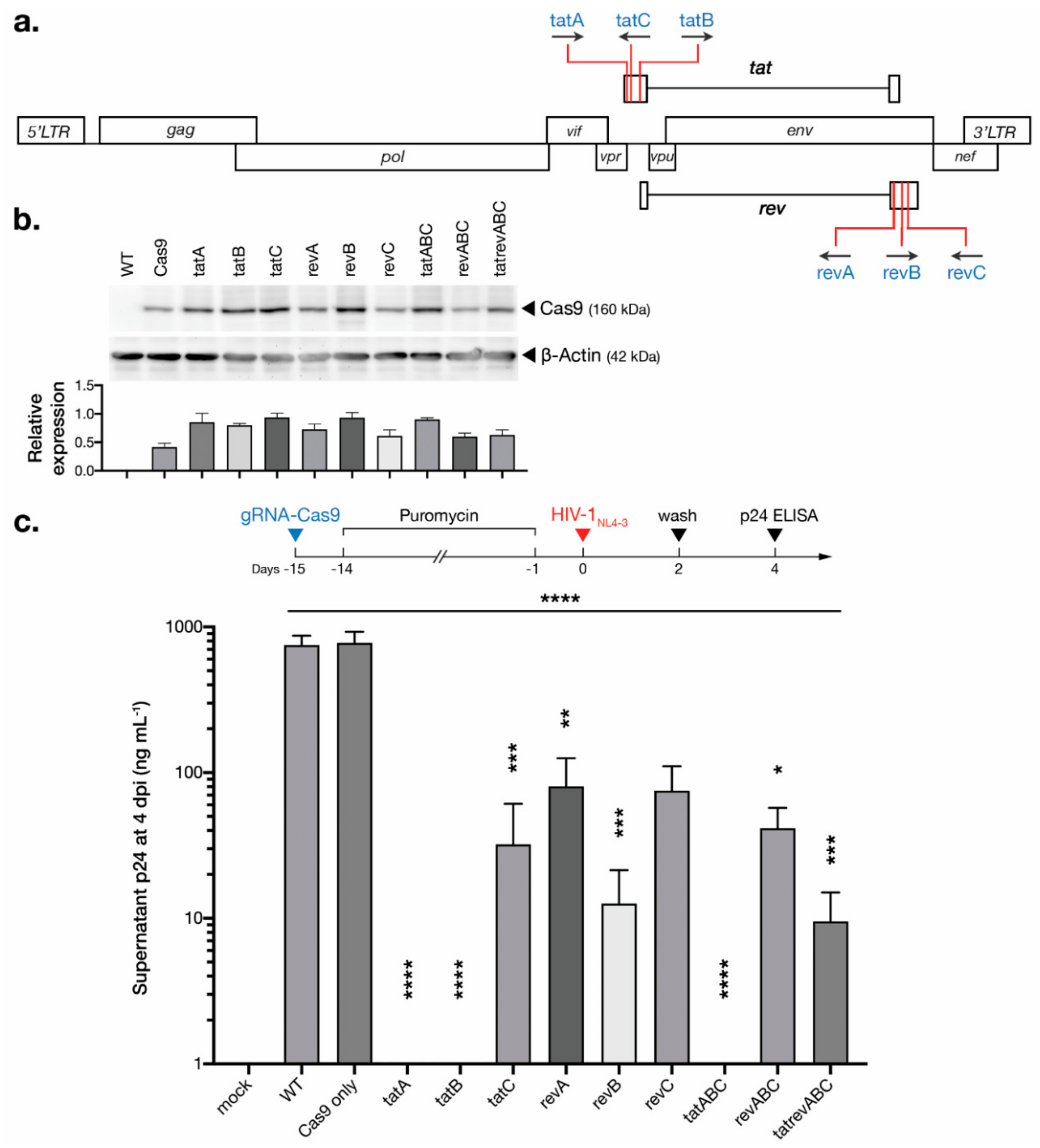
3.1. CRISPR-Cas9 Targeting tat and rev Protects T Cells from De Novo HIV-1 Infection
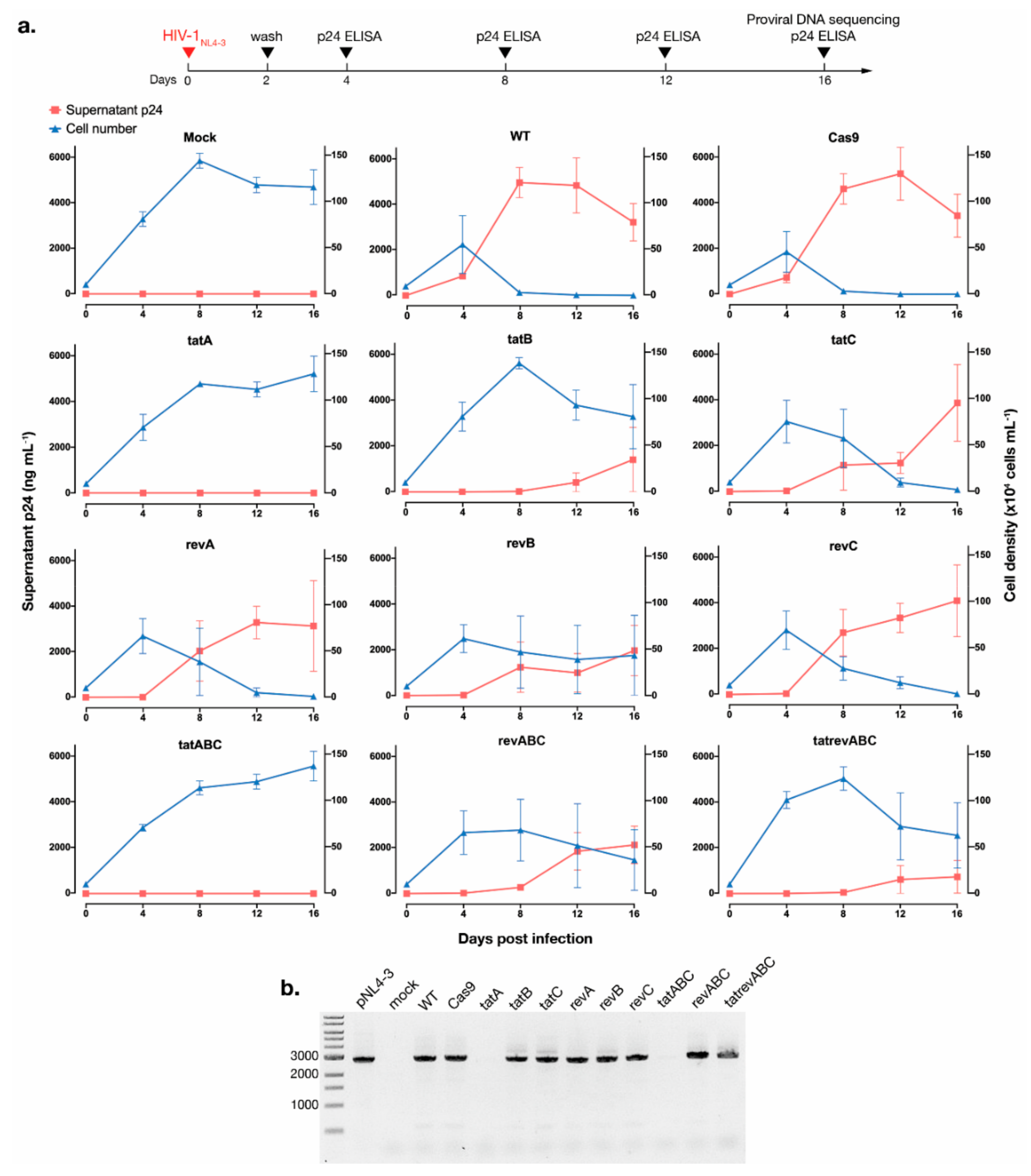
3.2. CRISPR-Cas9 Targeting tat Inhibited HIV-1 Escape Observed for All Other gRNAs
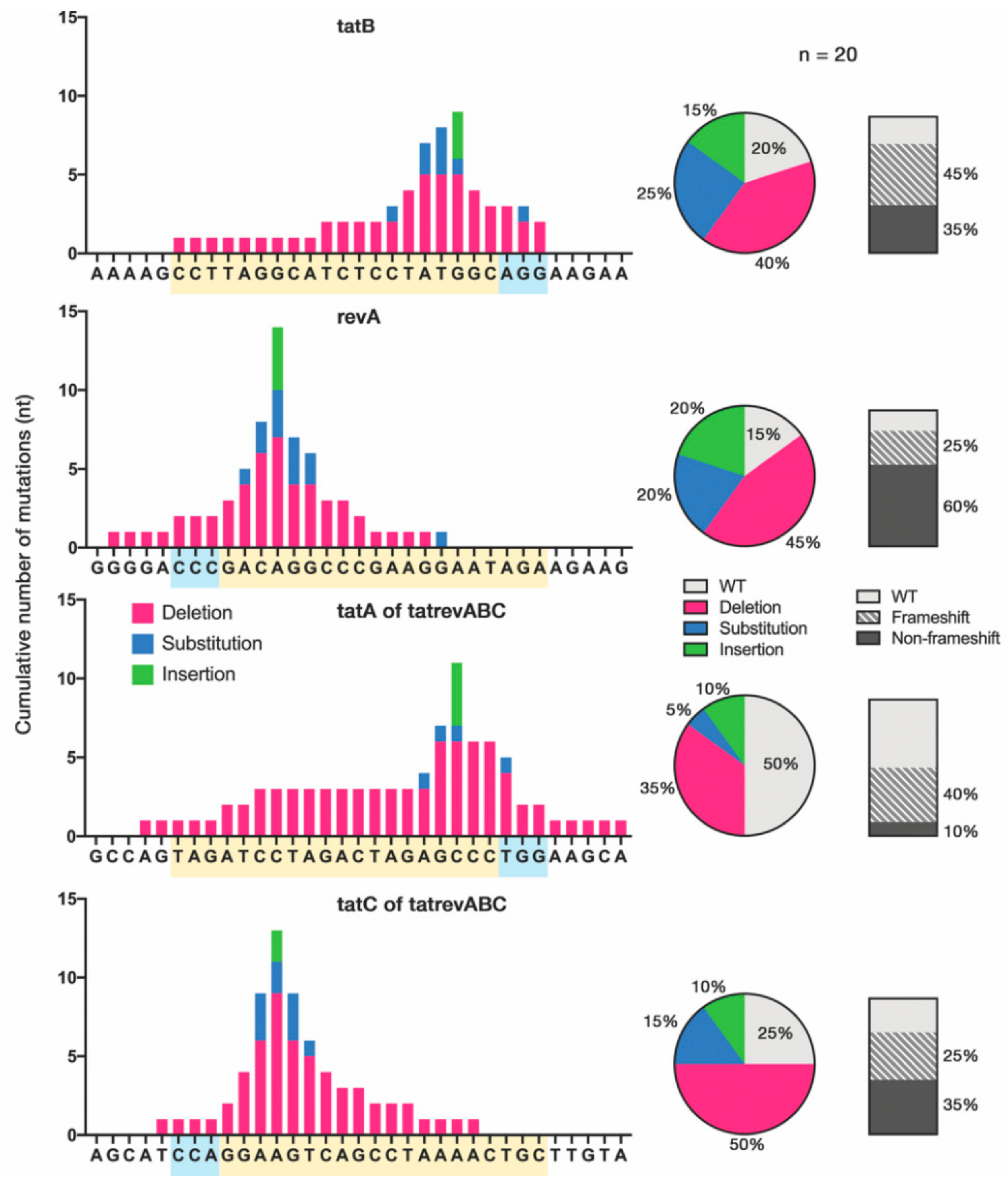
3.3. Cas9-Induced Mutational Pattern of HIV-1 Provirus after Prolonged Acute Infection
3.4. Cas9-tat gRNAs Maintained HIV-1 Suppression after 2nd Round Infection and Co-Culture with Unprotected T Cells
3.5. Development of Multiplexed tat/rev-Targeting CRISPR-Cas9 in an All-in-One Vector
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Das, A.T.; Binda, C.S.; Berkhout, B. Elimination of infectious HIV DNA by CRISPR–Cas9. Curr. Opin. Virol. 2019, 38, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Panfil, A.R.; London, J.A.; Green, P.L.; Yoder, K.E. CRISPR/Cas9 genome editing to disable the latent HIV-1 provirus. Front. Microbiol. 2018, 9, 3107. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Gao, Z.; Berkhout, B. CRISPR therapy towards an HIV cure. Brief. Funct. Genomics 2020, 19, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 2510. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Zhang, T.; Li, F.; Yang, F.; Putatunda, R.; Young, W.-B.; Khalili, K.; Hu, W.; Zhang, Y. Functional screening of guide RNAs targeting the regulatory and structural HIV-1 viral genome for a cure of AIDS. AIDS 2016, 30, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Yoder, K.E.; Bundschuh, R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/Cas9. Sci. Rep. 2016, 6, 29530. [Google Scholar] [CrossRef]
- Mefferd, A.L.; Bogerd, H.P.; Irwan, I.D.; Cullen, B.R. Insights into the mechanisms underlying the inactivation of HIV-1 proviruses by CRISPR/Cas. Virology 2018, 520, 116–126. [Google Scholar] [CrossRef]
- Canver, M.C.; Bauer, D.E.; Dass, A.; Yien, Y.Y.; Chung, J.; Masuda, T.; Maeda, T.; Paw, B.H.; Orkin, S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014, 289, 21312–21324. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–82. [Google Scholar] [CrossRef]
- Ophinni, Y.; Inoue, M.; Kotaki, T.; Kameoka, M. CRISPR/Cas9 system targeting regulatory genes of HIV-1 inhibits viral replication in infected T-cell cultures. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef]
- Liang, C.; Wainberg, M.A.; Das, A.T.; Berkhout, B. CRISPR/Cas9: A double-edged sword when used to combat HIV infection. Retrovirology 2016, 13, 37. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Ebina, H.; Kanemura, Y.; Misawa, N.; Koyanagi, Y. Anti-HIV-1 potency of the CRISPR/Cas9 system insufficient to fully inhibit viral replication. Microbiol. Immunol. 2016, 60, 483–496. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Cui, Y.C.; Pan, Q.; Zhu, W.; Gendron, P.; Guo, F.; Cen, S.; Witcher, M.; Liang, C. HIV-1 employs multiple mechanisms to resist Cas9/single guide RNA targeting the viral primer binding site. J. Virol. 2018, 92, e01135-18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, G.; Das, A.T.; Berkhout, B. Combinatorial CRISPR-Cas9 and RNA interference attack on HIV-1 DNA and RNA can lead to cross-resistance. Antimicrob. Agents Chemother. 2017, 61, e01486-17. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A combinatorial CRISPR-Cas9 attack on HIV-1 DNA extinguishes all infectious provirus in infected T cell cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Lebbink, R.J.; de Jong, D.C.M.; Wolters, F.; Kruse, E.M.; van Ham, P.M.; Wiertz, E.J.H.J.; Nijhuis, M. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci. Rep. 2017, 7, 41968. [Google Scholar] [CrossRef]
- Bella, R.; Kaminski, R.; Mancuso, P.; Young, W.-B.; Chen, C.; Sariyer, R.; Fischer, T.; Amini, S.; Ferrante, P.; Jacobson, J.M.; et al. Removal of HIV DNA by CRISPR from patient blood engrafts in humanized mice. Mol. Ther. Nucleic Acids 2018, 12, 275–282. [Google Scholar] [CrossRef]
- Roychoudhury, P.; De Silva Feelixge, H.; Reeves, D.; Mayer, B.T.; Stone, D.; Schiffer, J.T.; Jerome, K.R. Viral diversity is an obligate consideration in CRISPR/Cas9 designs for targeting the HIV reservoir. BMC Biol. 2018, 16, 75. [Google Scholar] [CrossRef]
- Darcis, G.; Binda, C.S.; Klaver, B.; Herrera-Carrillo, E.; Berkhout, B.; Das, A.T. The impact of HIV-1 genetic diversity on CRISPR-Cas9 antiviral activity and viral escape. Viruses 2019, 11, 255. [Google Scholar] [CrossRef]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K. HIV-1 Tat: Structure and function. In Human Retroviruses and AIDS 1996: A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences; US Department of Energy: Washington, DC, USA, 1996; pp. 11–26. [Google Scholar]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-and-lock strategies to cure HIV infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Sun, Y.; Li, D.; Lin, M.-H.; Lor, M.; Rustanti, L.; Harrich, D. Strong in vivo inhibition of HIV-1 replication by nullbasic, a tat mutant. MBio 2019, 10, e01769-19. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, R.A.M.; Meyer, B.E.; Simon, J.H.M.; Fischer, U.; Malim, M.H. HIV-1 infection of non-dividing cells: Evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J. 1997, 16, 4531–4539. [Google Scholar] [CrossRef]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of interferon-stimulated genes with antiretroviral activity. Cell Host Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef]
- Traxler, E.A.; Yao, Y.; Wang, Y.-D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016, 22, 987–990. [Google Scholar] [CrossRef]
- Pirona, A.C.; Oktriani, R.; Boettcher, M.; Hoheisel, J.D. Process for an efficient lentiviral cell transduction. Biol. Methods Protoc. 2020, 5, bpaa005. [Google Scholar] [CrossRef]
- Harada, S.; Koyanagi, Y.; Yamamoto, N. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 1985, 229, 563 LP–566. [Google Scholar] [CrossRef]
- Fernandez, M.V.; Delviks-Frankenberry, K.A.; Scheiblin, D.A.; Happel, C.; Pathak, V.K.; Freed, E.O. Authentication analysis of MT-4 cells distributed by the National Institutes of Health AIDS Reagent Program. J. Virol. 2019, 93, e01390-19. [Google Scholar] [CrossRef]
- Calabrò, M.L.; Zanotto, C.; Calderazzo, F.; Crivellaro, C.; Del Mistro, A.; de Rossi, A.; Chieco-Bianchi, L. HIV-1 infection of the thymus: Evidence for a cytopathic and thymotropic viral variant in vivo. AIDS Res. Hum. Retroviruses 1995, 11, 11–19. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- McCarty, N.S.; Graham, A.E.; Studená, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef]
- Deyle, D.R.; Russell, D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther. 2009, 11, 442–447. [Google Scholar] [PubMed]
- ter Brake, O.; Hooft, K.; Liu, Y.P.; Centlivre, M.; von Eije, K.J.; Berkhout, B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol. Ther. 2008, 16, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Huang, C.; Liang, Z.; Ma, X.; Wang, N.; Huo, Y.-X. Reversed paired-gRNA plasmid cloning strategy for efficient genome editing in Escherichia coli. Microb. Cell Fact. 2020, 19, 63. [Google Scholar] [CrossRef]
- De Silva Feelixge, H.S.; Stone, D.; Pietz, H.L.; Roychoudhury, P.; Greninger, A.L.; Schiffer, J.T.; Aubert, M.; Jerome, K.R. Detection of treatment-resistant infectious HIV after genome-directed antiviral endonuclease therapy. Antiviral Res. 2016, 126, 90–98. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schiffer, J.T.; Aubert, M.; Weber, N.D.; Mintzer, E.; Stone, D.; Jerome, K.R. Targeted DNA mutagenesis for the cure of chronic viral infections. J. Virol. 2012, 86, 8920–8936. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.S.; Klaver, B.; Berkhout, B.; Das, A.T. CRISPR-Cas9 dual-gRNA attack causes mutation, excision and inversion of the HIV-1 proviral DNA. Viruses 2020, 12, 330. [Google Scholar] [CrossRef]
- Chung, C.H.; Allen, A.G.; Sullivan, N.T.; Atkins, A.; Nonnemacher, M.R.; Wigdahl, B.; Dampier, W. Computational analysis concerning the impact of DNA accessibility on CRISPR-Cas9 cleavage efficiency. Mol. Ther. 2019, 28, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Link, R.W.; Nonnemacher, M.R.; Wigdahl, B.; Dampier, W. Prediction of human immunodeficiency virus type 1 subtype-specific off-target effects arising from CRISPR-Cas9 gene editing therapy. Cris. J. 2018, 1, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Adikusuma, F.; Piltz, S.; Corbett, M.A.; Turvey, M.; McColl, S.R.; Helbig, K.J.; Beard, M.R.; Hughes, J.; Pomerantz, R.T.; Thomas, P.Q. Large deletions induced by Cas9 cleavage. Nature 2018, 560, E8–E9. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Allen, A.G.; Chung, C.-H.; Atkins, A.; Dampier, W.; Khalili, K.; Nonnemacher, M.R.; Wigdahl, B. Gene editing of HIV-1 co-receptors to prevent and/or cure virus infection. Front. Microbiol. 2018, 9, 2940. [Google Scholar] [CrossRef]
- Dampier, W.; Nonnemacher, M.R.; Sullivan, N.T.; Jacobson, J.M.; Wigdahl, B. HIV excision utilizing CRISPR/Cas9 technology: Attacking the proviral quasispecies in reservoirs to achieve a cure. MOJ Immunol. 2014, 1, 00022. [Google Scholar] [CrossRef] [PubMed]
- Dampier, W.; Sullivan, N.T.; Mell, J.C.; Pirrone, V.; Ehrlich, G.D.; Chung, C.H.; Allen, A.G.; DeSimone, M.; Zhong, W.; Kercher, K.; et al. Broad-spectrum and personalized guide RNAs for CRISPR/Cas9 HIV-1 therapeutics. AIDS Res. Hum. Retroviruses 2018, 34, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Spector, C.; Mele, A.R.; Wigdahl, B.; Nonnemacher, M.R. Genetic variation and function of the HIV-1 Tat protein. Med. Microbiol. Immunol. 2019, 208, 131–169. [Google Scholar] [CrossRef]
- Walter, D.M.; Venancio, O.S.; Buza, E.L.; Tobias, J.W.; Deshpande, C.; Gudiel, A.A.; Kim-Kiselak, C.; Cicchini, M.; Yates, T.J.; Feldser, D.M. Systematic in vivo inactivation of chromatin-regulating enzymes identifies Setd2 as a potent tumor suppressor in lung adenocarcinoma. Cancer Res. 2017, 77, 1719–1729. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, S.; Seo, J.H.; Choi, J.W.; Park, J.; Min, S.; Yoon, S.; Cho, S.-R.; Kim, H.H. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, S.; Liu, Z.; Ke, Z.; Li, C.; Yu, X.; Chen, S.; Guo, D. Genome scale screening identification of SaCas9/gRNAs for targeting HIV-1 provirus and suppression of HIV-1 infection. Virus Res. 2018, 250, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vidigal, J.A.; Ventura, A. Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun. 2015, 6, 8083. [Google Scholar] [CrossRef] [PubMed]
- Kabadi, A.M.; Ousterout, D.G.; Hilton, I.B.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014, 42, e147. [Google Scholar] [CrossRef]
- Xie, K.; Minkenberg, B.; Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. 2015, 112, 3570 LP–3575. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Skrekas, C.; Nielsen, J.; David, F. Multiplexed CRISPR/Cas9 genome editing and gene regulation using Csy4 in Saccharomyces cerevisiae. ACS Synth. Biol. 2018, 7, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef]
- Gao, Z.; Herrera-Carrillo, E.; Berkhout, B. Improvement of the CRISPR-Cpf1 system with ribozyme-processed crRNA. RNA Biol. 2018, 15, 1458–1467. [Google Scholar] [CrossRef]
- Gao, Z.; Fan, M.; Das, A.T.; Herrera-Carrillo, E.; Berkhout, B. Extinction of all infectious HIV in cell culture by the CRISPR-Cas12a system with only a single crRNA. Nucleic Acids Res. 2020, 48, 5527–5539. [Google Scholar] [CrossRef]
- Kaminski, R.; Chen, Y.; Salkind, J.; Bella, R.; Young, W.-B.; Ferrante, P.; Karn, J.; Malcolm, T.; Hu, W.; Khalili, K. Negative feedback regulation of HIV-1 by gene editing strategy. Sci. Rep. 2016, 6, 31527. [Google Scholar] [CrossRef]
- Vergara-Mendoza, M.; Gomez-Quiroz, L.E.; Miranda-Labra, R.U.; Fuentes-Romero, L.L.; Romero-Rodríguez, D.P.; González-Ruiz, J.; Hernández-Rizo, S.; Viveros-Rogel, M. Regulation of Cas9 by viral proteins Tat and Rev for HIV-1 inactivation. Antiviral Res. 2020, 180, 104856. [Google Scholar] [CrossRef]
- Kaushik, A.; Yndart, A.; Atluri, V.; Tiwari, S.; Tomitaka, A.; Gupta, P.; Jayant, R.D.; Alvarez-Carbonell, D.; Khalili, K.; Nair, M. Magnetically guided non-invasive CRISPR-Cas9/gRNA delivery across blood-brain barrier to eradicate latent HIV-1 infection. Sci. Rep. 2019, 9, 3928. [Google Scholar] [CrossRef]
- Campbell, L.A.; Coke, L.M.; Richie, C.T.; Fortuno, L.V.; Park, A.Y.; Harvey, B.K. Gesicle-mediated delivery of CRISPR/Cas9 ribonucleoprotein complex for inactivating the HIV provirus. Mol. Ther. 2019, 27, 151–163. [Google Scholar] [CrossRef]
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gRNA | Target Sequence + PAM a | Position in HXB2 | Shannon Entropy b | Specificity c | Knock-out Efficiency c | MMEJ deletion c | Frameshiftc |
|---|---|---|---|---|---|---|---|
| tatA | TAGATCCTAGACTAGAGCCCTGG | 5840–5862 | 0.14 | 76 | 39 | 61 | 77 |
| tatB | CCTTAGGCATCTCCTATGGCAGG | 5954–5976 | 0.06 | 91 | 53 | 89 | 74 |
| tatC | CCAGGAAGTCAGCCTAAAACTGC | 5891–5869 | 0.22 | 81 | 35 | 58 | 79 |
| revA | CCCGACAGGCCCGAAGGAATAGA | 8415–8393 | 0.24 | 94 | 38 | 77 | 83 |
| revB | CACTTATCTGGGACGATCTGCGG | 8475–8497 | 0.32 | 95 | 63 | 66 | 87 |
| revC | CCACCGCTTGAGAGACTTACTCT | 8540–8518 | 0.25 | 94 | 63 | 37 | 64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ophinni, Y.; Miki, S.; Hayashi, Y.; Kameoka, M. Multiplexed tat-Targeting CRISPR-Cas9 Protects T Cells from Acute HIV-1 Infection with Inhibition of Viral Escape. Viruses 2020, 12, 1223. https://doi.org/10.3390/v12111223
Ophinni Y, Miki S, Hayashi Y, Kameoka M. Multiplexed tat-Targeting CRISPR-Cas9 Protects T Cells from Acute HIV-1 Infection with Inhibition of Viral Escape. Viruses. 2020; 12(11):1223. https://doi.org/10.3390/v12111223
Chicago/Turabian StyleOphinni, Youdiil, Sayaka Miki, Yoshitake Hayashi, and Masanori Kameoka. 2020. "Multiplexed tat-Targeting CRISPR-Cas9 Protects T Cells from Acute HIV-1 Infection with Inhibition of Viral Escape" Viruses 12, no. 11: 1223. https://doi.org/10.3390/v12111223
APA StyleOphinni, Y., Miki, S., Hayashi, Y., & Kameoka, M. (2020). Multiplexed tat-Targeting CRISPR-Cas9 Protects T Cells from Acute HIV-1 Infection with Inhibition of Viral Escape. Viruses, 12(11), 1223. https://doi.org/10.3390/v12111223