
Advances in the Bioinformatics Knowledge of mRNA Polyadenylation in Baculovirus Genes

 , ,
, , 
Abstract
:1. Introduction
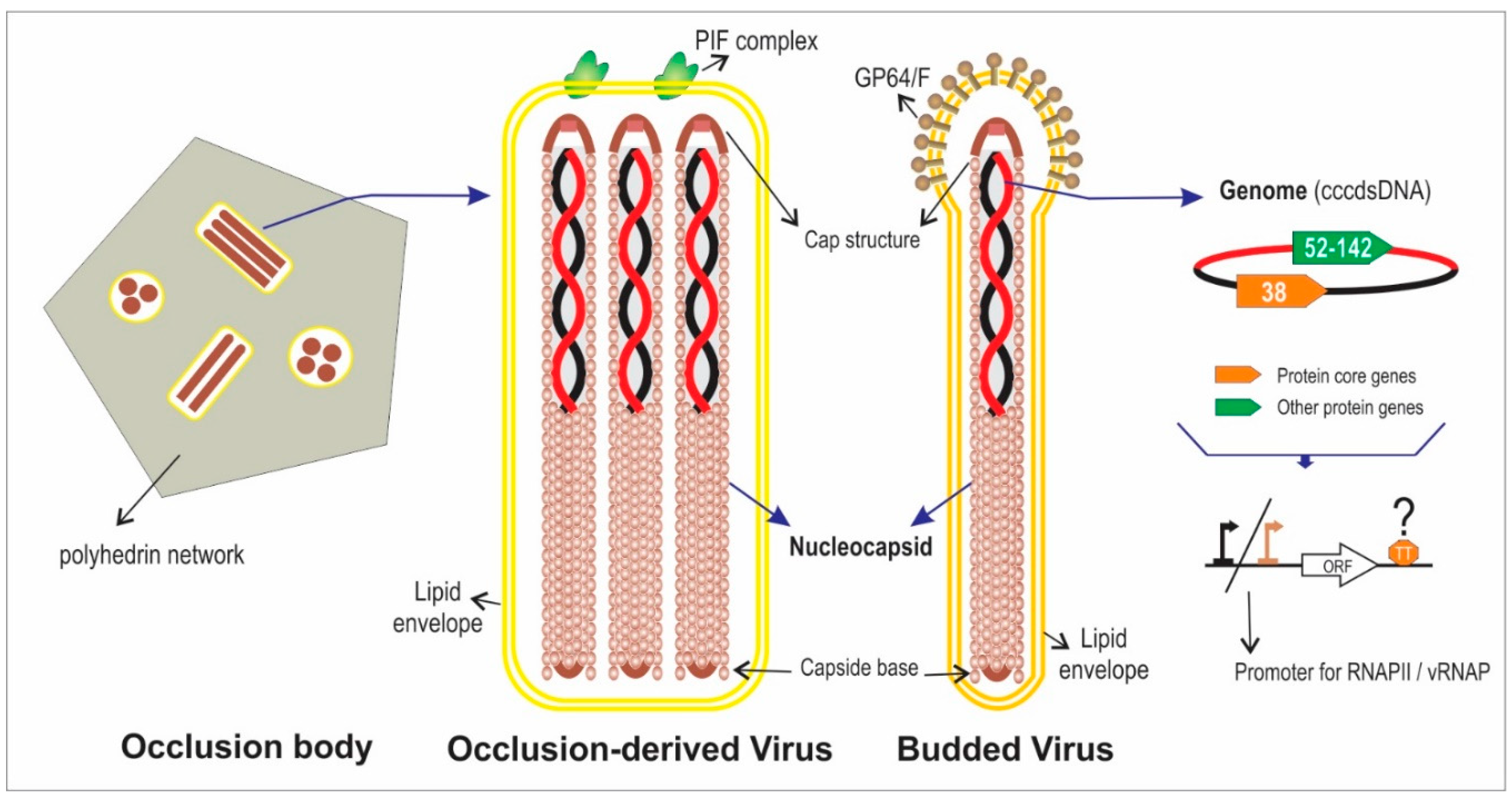
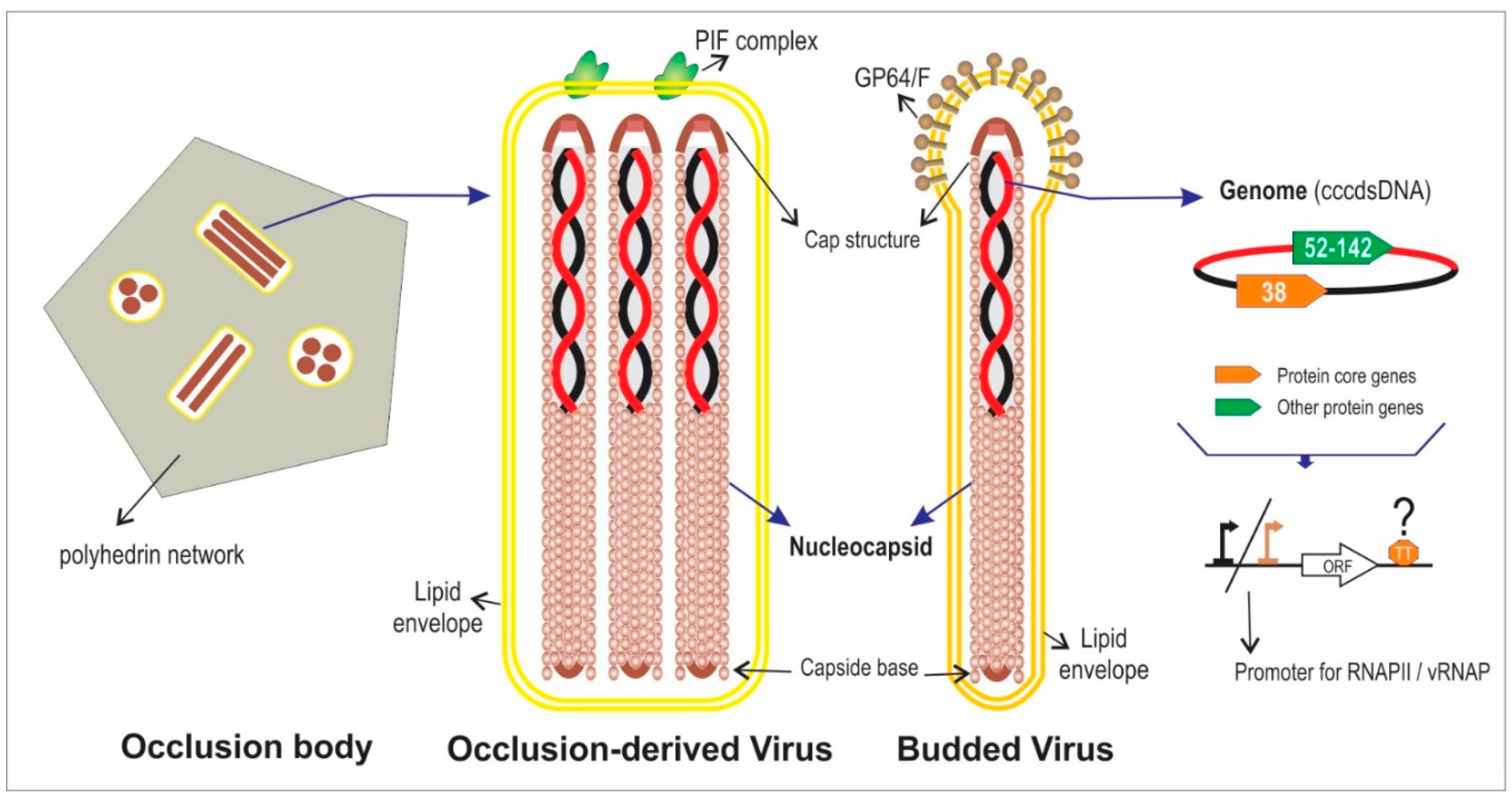
2. Actual Genomic Knowledge about the Family Baculoviridae
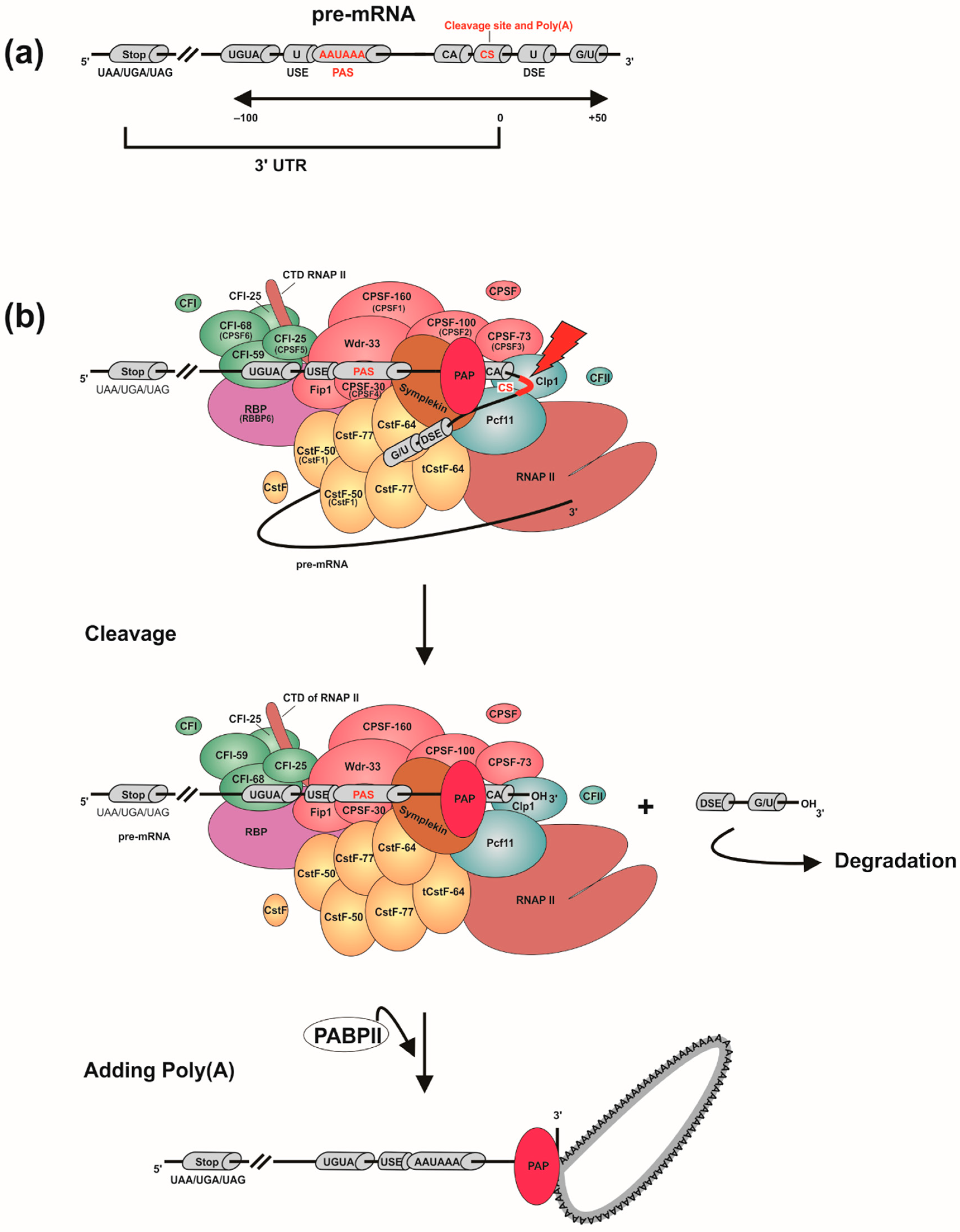
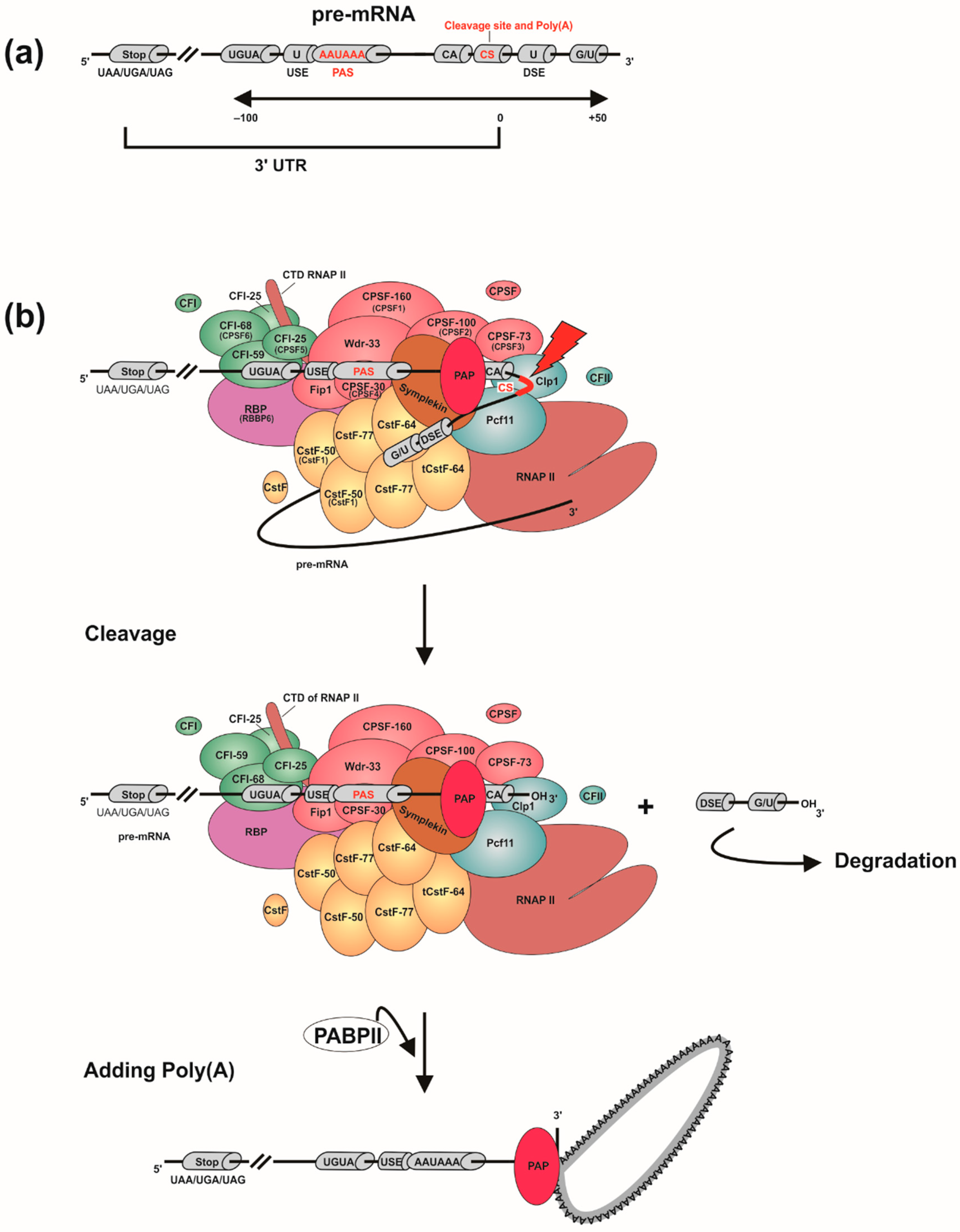
3. Eukaryotic mRNA 3′ End Processing
4. How Bioinformatics Can Assist in the Characterization of the Baculoviral mRNA 3′ End Processing
4.1. Bioinformatic Detection of Signals Involved in the Processing of the 3′ End of Genes
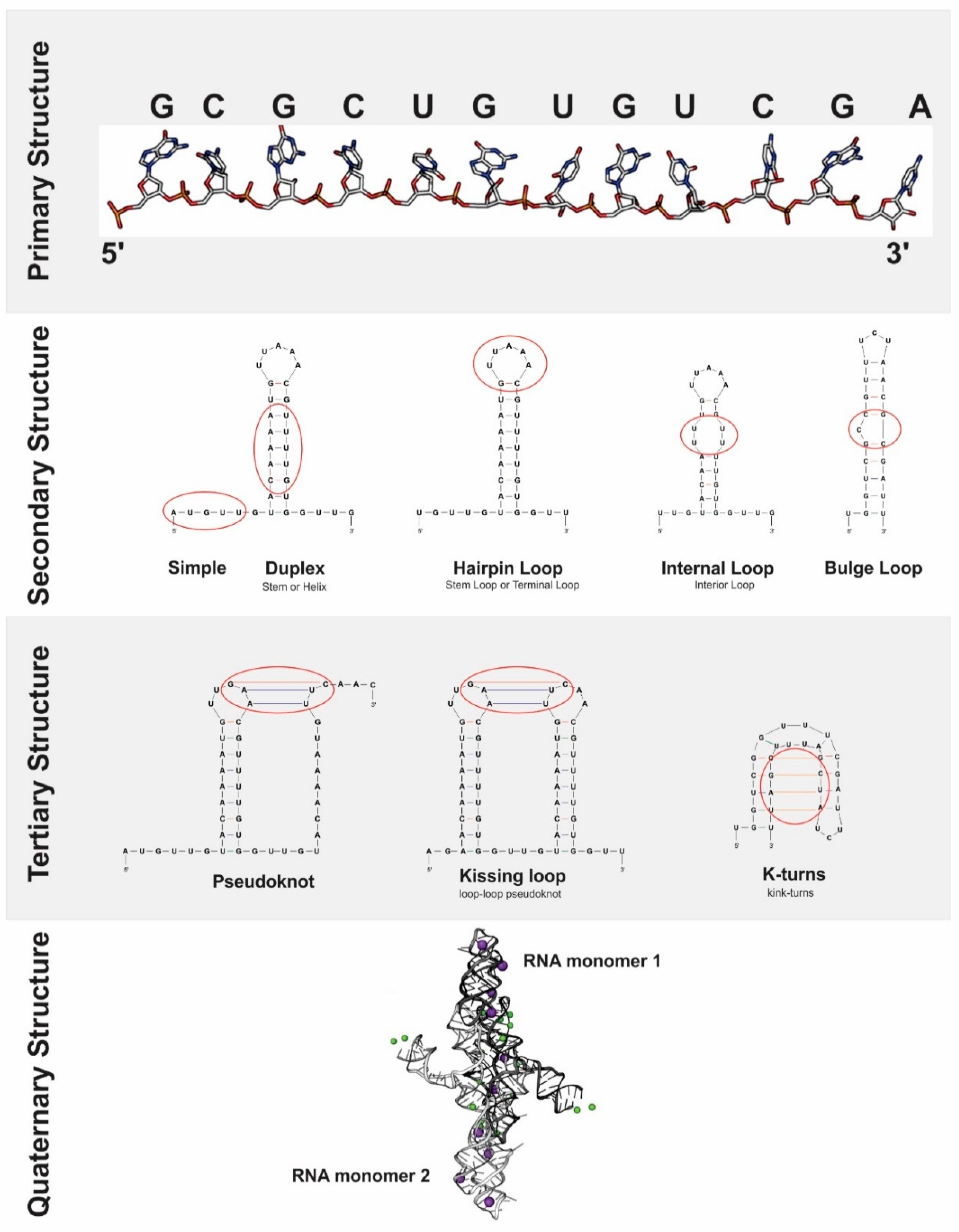
4.2. Bioinformatic Prediction of RNA Structure
5. Baculoviral mRNA 3′ End Processing
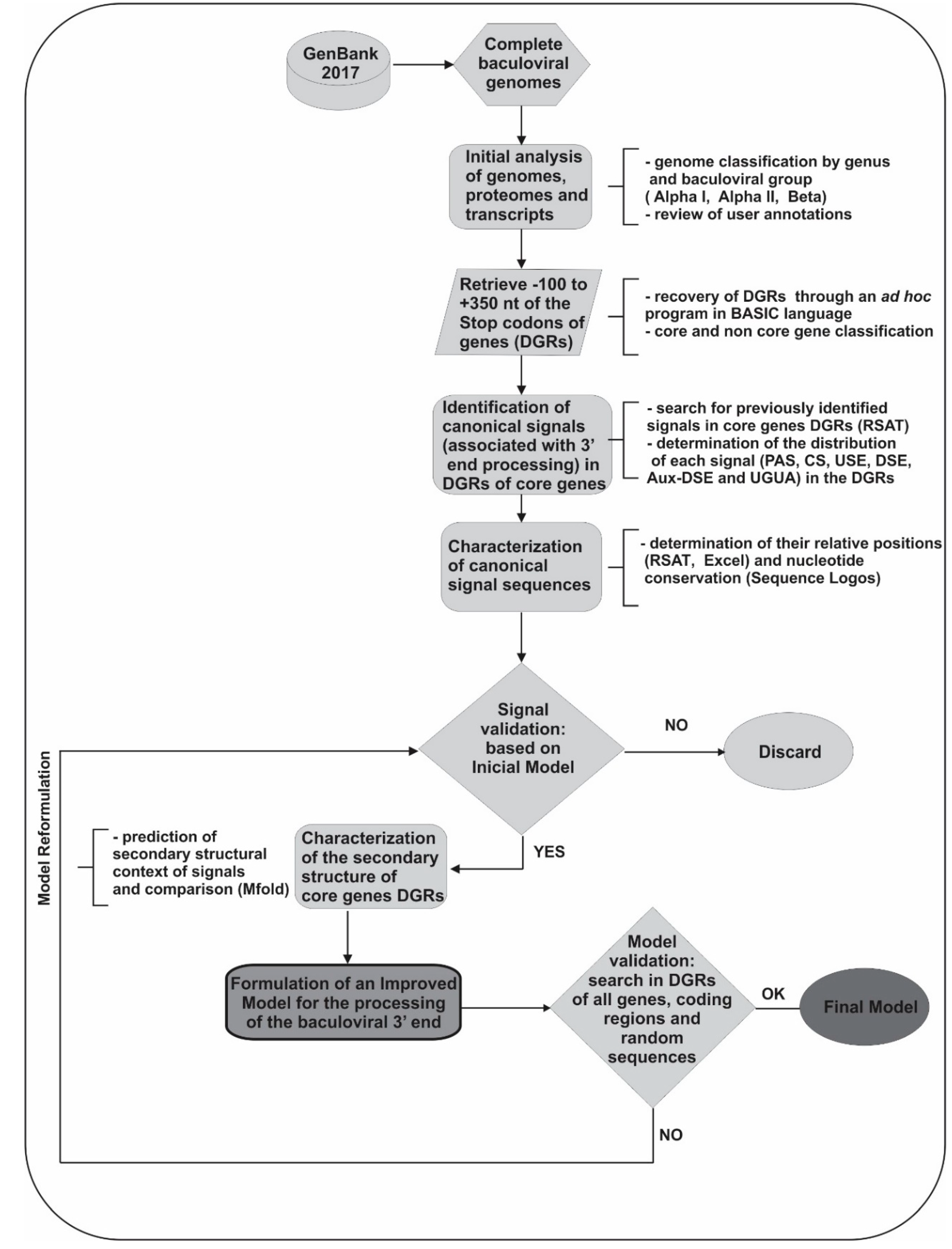
5.1. Bibliographic Data Mining
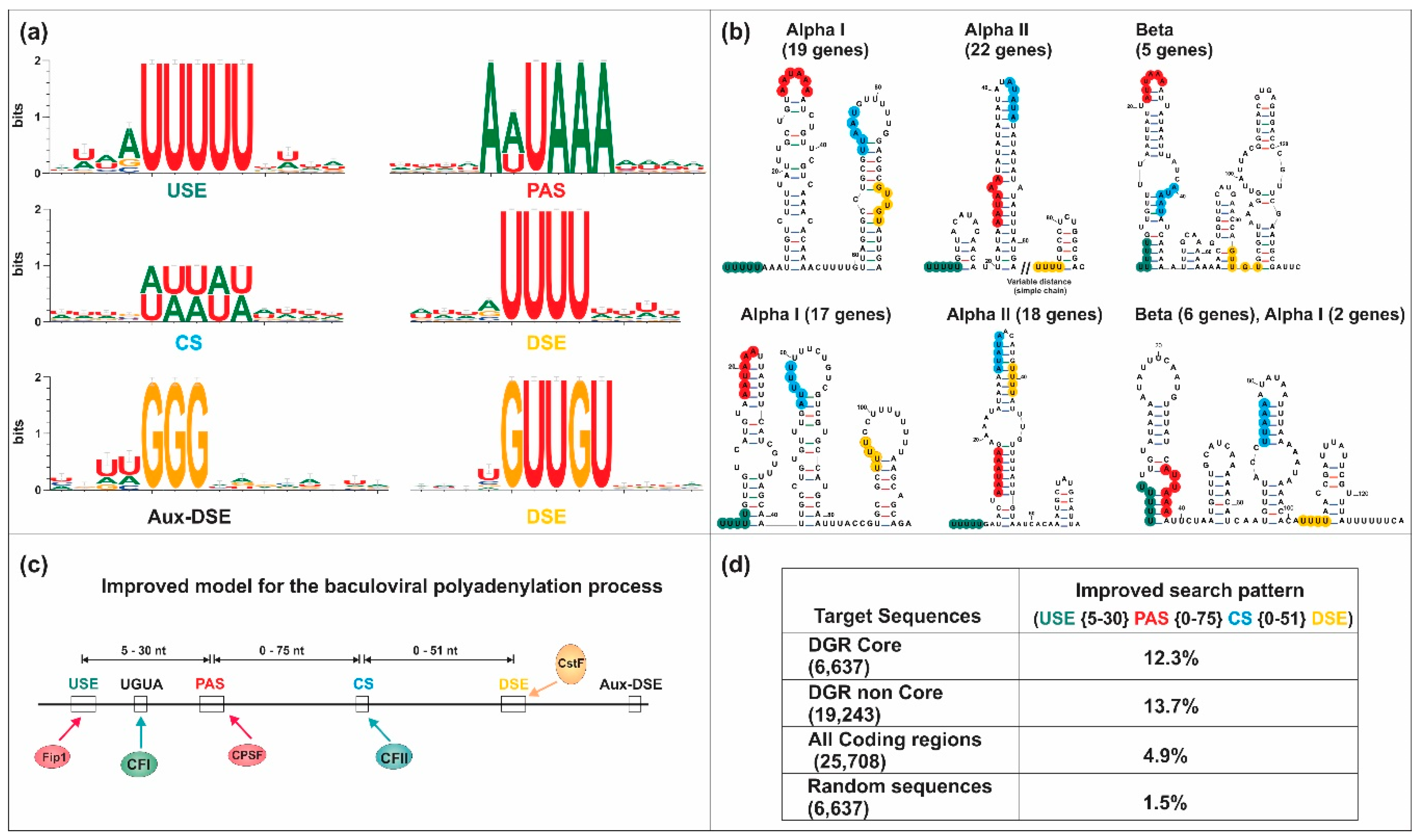
5.2. Bioinformatic Analyses
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Klasberg, S.; Bitard-Feildel, T.; Mallet, L. Computational identification of novel genes: Current and future perspectives. Bioinform. Biol. Insights 2016, 10, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemann, S.; Arlt, D.; Huber, W.; Wellenreuther, R.; Schleeger, S.; Mehrle, A.; Bechtel, S.; Sauermann, M.; Korf, U.; Pepperkok, R.; et al. From ORFeome to biology: A functional genomics pipeline. Genome Res. 2004, 14, 2136–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Down, T.A.; Hubbard, T.J. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 2002, 12, 458–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnevsky, O.V.; Kolchanov, N.A. ARGO: A web system for the detection of degenerate motifs and large-scale recognition of eukaryotic promoters. Nucleic Acids Res. 2005, 33, W417–W422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.E.; Pattison, A.D.; Beilharz, T.H. Alternative polyadenylation in the regulation and dysregulation of gene expression. Semin. Cell. Dev. Biol. 2018, 75, 61–69. [Google Scholar] [CrossRef]
- Miele, S.A.B.; Belaich, M.N.; Ghiringhelli, P.D. Quality control of baculoviral bioinsecticide production. In Wide Spectra in Quality Control; Akyar, I., Ed.; IntechOpen: London, UK, 2011; pp. 411–428. ISBN 978-953-307-683-6. [Google Scholar] [CrossRef]
- Popham, H.J.; Nusawardani, T.; Bonning, B.C. Introduction to the use of baculoviruses as biological insecticides. Methods Mol. Biol. 2016, 1350, 383–392. [Google Scholar] [CrossRef]
- López, M.G.; Diez, M.; Alfonso, V.; Taboga, O. Biotechnological applications of occlusion bodies of Baculoviruses. Appl. Microbiol. Biotechnol. 2018, 102, 6765–6774. [Google Scholar] [CrossRef]
- Possee, R.D.; Chambers, A.C.; Graves, L.P.; Aksular, M.; King, L.A. Recent developments in the use of baculovirus expression vectors. Curr. Issues Mol. Biol. 2020, 34, 215–230. [Google Scholar] [CrossRef]
- Tsai, C.H.; Wei, S.C.; Lo, H.R.; Chao, Y.C. Baculovirus as versatile vectors for protein display and biotechnological applications. Curr. Issues Mol. Biol. 2020, 34, 231–256. [Google Scholar] [CrossRef] [Green Version]
- Rohrmann, G.F. Baculovirus Molecular Biology, 4th ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2019.
- Herniou, E.A.; Luque, T.; Chen, X.; Vlak, J.M.; Winstanley, D.; Cory, J.S.; O’Reilly, D.R. Use of whole genome sequence data to infer baculovirus phylogeny. J. Virol. 2001, 75, 8117–8126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herniou, E.A.; Olszewski, J.A.; Cory, J.S.; O’Reilly, D.R. The genome sequence and evolution of baculoviruses. Annu. Rev. Entomol. 2003, 48, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Blissard, G.W.; Bonning, B.C.; Cory, J.S.; Herniou, E.A.; Rohrmann, G.F.; Theilmann, D.A.; Thiem, S.M.; Vlak, J.M. On the classification and nomenclature of baculoviruses: A proposal for revision. Arch. Virol. 2006, 151, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.N.; Rohrmann, G.F. Transfer, incorporation, and substitution of envelope fusion proteins among members of the Baculoviridae, Orthomyxoviridae, and Metaviridae (insect retrovirus) families. J. Virol. 2002, 76, 5301–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, S.A.B.; Garavaglia, M.J.; Belaich, M.N.; Ghiringhelli, P.D. Baculovirus: Molecular insights on their diversity and conservation. Int. J. Evol. Biol. 2011, 2011, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garavaglia, M.J.; Miele, S.A.B.; Iserte, J.A.; Belaich, M.N.; Ghiringhelli, P.D. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 2012, 86, 12069–12079. [Google Scholar] [CrossRef] [Green Version]
- Javed, M.A.; Biswas, S.; Willis, L.G.; Harris, S.; Pritchard, C.; van Oers, M.M.; Donly, B.C.; Erlandson, M.A.; Hegedus, D.D.; Theilmann, D.A. Autographa californica Multiple Nucleopolyhedrovirus AC83 is a per os infectivity factor (PIF) protein required for occlusion-derived virus (ODV) and budded virus nucleocapsid assembly as well as assembly of the PIF complex in ODV envelopes. J. Virol. 2017, 91, e02115–e02116. [Google Scholar] [CrossRef] [Green Version]
- Thézé, J.; Lopez-Vaamonde, C.; Cory, J.S.; Herniou, E.A. Biodiversity, evolution and ecological specialization of baculoviruses: A treasure trove for future applied research. Viruses 2018, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Ferrelli, M.L.; Berretta, M.F.; Belaich, M.N.; Ghiringhelli, P.D.; Sciocco-Cap, A.; Romanowski, V. The baculoviral genome. In Viral Genomes—Molecular Structure, Diversity, Gene Expression Mechanisms and Host-Virus Interactions; Garcia, M.L., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0098-0. [Google Scholar] [CrossRef]
- De Vries, H.; Rüegsegger, U.; Hübner, W.; Friedlein, A.; Langen, H.; Keller, W. Human pre-mRNA cleavage factor II(m) contains homologs of yeast proteins and bridges two other cleavage factors. EMBO J. 2000, 19, 5895–5904. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Salinas, C.A.; Sinclair, D.A.; O’Hare, K.; Brock, H.W. Characterization of a Drosophila homologue of the 160-kDa subunit of the cleavage and polyadenylation specificity factor CPSF. Mol. Gen. Genet. 1998, 257, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Lu, J.; Kulbokas, E.J.; Golub, T.R.; Mootha, V.; Lindblad-Toh, K.; Lander, E.S.; Kellis, M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 2005, 434, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.S.; Yong, J. Alternative polyadenylation of mRNAs: 3′-untranslated region matters in gene expression. Mol. Cells. 2016, 39, 281–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataraman, K.; Brown, K.M.; Gilmartin, G.M. Analysis of a noncanonical poly(A) site reveals a tripartite mechanism for vertebrate poly(A) site recognition. Genes Dev. 2005, 19, 1315–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalziel, M.; Nunes, N.M.; Furger, A. Two G-rich regulatory elements located adjacent to and 440 nucleotides downstream of the core poly(A) site of the intronless melanocortin receptor 1 gene are critical for efficient 3′ end processing. Mol. Cell Biol. 2007, 27, 1568–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatton, L.S.; Eloranta, J.J.; Figueiredo, L.M.; Takagaki, Y.; Manley, J.L.; O’Hare, K. The Drosophila homologue of the 64 kDa subunit of cleavage stimulation factor interacts with the 77 kDa subunit encoded by the suppressor of forked gene. Nucleic Acids Res. 2000, 28, 520–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, U.; Gündel, M.; Knoth, A.; Kerwitz, Y.; Rüdel, S.; Wahle, E. Poly(A) tail length is controlled by the nuclear poly(A)-binding protein regulating the interaction between poly(A) polymerase and the cleavage and polyadenylation specificity factor. J. Biol. Chem. 2009, 284, 22803–22814. [Google Scholar] [CrossRef] [Green Version]
- Parker, R.; Sheth, U. P bodies and the control of mRNA translation and degradation. Mol. Cell. 2007, 25, 635–646. [Google Scholar] [CrossRef]
- Proudfoot, N.; O’Sullivan, J. Polyadenylation: A tail of two complexes. Curr. Biol. 2002, 12, R855–R857. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Manley, J.L. Alternative cleavage and polyadenylation: The long and short of it. Trends Biochem. Sci. 2013, 38, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.; Kyriakopoulou, C.B.; Virtanen, A. Identification of a stem-loop structure important for polyadenylation at the murine IgM secretory poly(A) site. Nucleic Acids Res. 1999, 27, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, M.J.; Daga, R.R.; Garzón, A.; Thode, G.; Jimenez, J. Poly(A) site choice during mRNA 3′-end formation in the Schizosaccharomyces pombe wos2 gene. Mol. Genet. Genomics. 2002, 267, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Huang, L.F.; Su, H.C.; Jeng, S.T. Effects of the multiple polyadenylation signal AAUAAA on mRNA 3′-end formation and gene expression. Planta 2009, 230, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Flomen, R.; Makoff, A. Increased RNA editing in EAAT2 pre-mRNA from amyotrophic lateral sclerosis patients: Involvement of a cryptic polyadenylation site. Neurosci. Lett. 2011, 497, 139–143. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef]
- Ji, Z.; Luo, W.; Li, W.; Hoque, M.; Pan, Z.; Zhao, Y.; Tian, B. Transcriptional activity regulates alternative cleavage and polyadenylation. Mol. Syst. Biol. 2011, 7, 534. [Google Scholar] [CrossRef]
- Takagaki, Y.; Seipelt, R.L.; Peterson, M.L.; Manley, J.L. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell 1996, 87, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol. 2012, 9, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Elkon, R.; Drost, J.; van Haaften, G.; Jenal, M.; Schrier, M.; Oude Vrielink, J.A.; Agami, R. E2F mediates enhanced alternative polyadenylation in proliferation. Genome Biol. 2012, 13, R59. [Google Scholar] [CrossRef] [Green Version]
- Yonaha, M.; Proudfoot, N.J. Specific transcriptional pausing activates polyadenylation in a coupled in vitro system. Mol. Cell. 1999, 3, 593–600. [Google Scholar] [CrossRef]
- Zarudnaya, M.I.; Kolomiets, I.M.; Potyahaylo, A.L.; Hovorun, D.M. Downstream elements of mammalian pre-mRNA polyadenylation signals: Primary, secondary and higher-order structures. Nucleic Acids Res. 2003, 31, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Martincic, K.; Alkan, S.A.; Cheatle, A.; Borghesi, L.; Milcarek, C. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat. Immunol. 2009, 10, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; Hoque, M.; Hou, L.; Shen, S.; Tian, B.; Dynlacht, B.D. PAF complex plays novel subunit-specific roles in alternative cleavage and polyadenylation. PLoS Genet. 2016, 12, e1005794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, S.I.; Polycarpou-Schwarz, M.; Mattaj, I.W. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol. Cell. 1998, 1, 255–264. [Google Scholar] [CrossRef]
- Millevoi, S.; Loulergue, C.; Dettwiler, S.; Karaa, S.Z.; Keller, W.; Antoniou, M.; Vagner, S. An interaction between U2AF 65 and CF I(m) links the splicing and 3’ end processing machineries. EMBO J. 2006, 25, 4854–4864. [Google Scholar] [CrossRef] [Green Version]
- Bava, F.A.; Eliscovich, C.; Ferreira, P.G.; Miñana, B.; Ben-Dov, C.; Guigó, R.; Valcárcel, J.; Méndez, R. CPEB1 coordinates alternative 3′-UTR formation with translational regulation. Nature 2013, 495, 121–125. [Google Scholar] [CrossRef]
- Jenal, M.; Elkon, R.; Loayza-Puch, F.; van Haaften, G.; Kühn, U.; Menzies, F.M.; Oude Vrielink, J.A.; Bos, A.J.; Drost, J.; Rooijers, K.; et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 2012, 149, 538–553. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; You, B.; Hoque, M.; Zheng, D.; Luo, W.; Ji, Z.; Park, J.Y.; Gunderson, S.I.; Kalsotra, A.; Manley, J.L.; et al. Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015, 11, e1005166. [Google Scholar] [CrossRef]
- Li, W.; Laishram, R.S.; Ji, Z.; Barlow, C.A.; Tian, B.; Anderson, R.A. Star-PAP control of BIK expression and apoptosis is regulated by nuclear PIPKIα and PKCδ signaling. Mol. Cell. 2012, 45, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.W.; Zhang, W.; Yeh, H.S.; de Jong, E.P.; Jun, S.; Kim, K.H.; Bae, S.S.; Beckman, K.; Hwang, T.H.; Kim, K.S.; et al. mRNA 3’-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015, 6, 7218. [Google Scholar] [CrossRef] [Green Version]
- Taub, F.E.; DeLeo, J.M.; Thompson, E.B. Sequential comparative hybridizations analyzed by computerized image processing can identify and quantitate regulated RNAs. DNA 1983, 2, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, P.J.; Choi, E.A.; Lu, J.; Flanagan, L.A.; Hertel, K.J.; Shi, Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 2011, 17, 761–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Sun, Y.; Li, Y.; Li, J.; Rao, X.; Chen, C.; Xu, A. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011, 21, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Jan, C.H.; Friedman, R.C.; Ruby, J.G.; Bartel, D.P. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 2011, 469, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Alternative polyadenylation: New insights from global analyses. RNA 2012, 18, 2105–2117. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.; Ji, Z.; Zheng, D.; Luo, W.; Li, W.; You, B.; Park, J.Y.; Yehia, G.; Tian, B. Analysis of alternative cleavage and polyadenylation by 3’ region extraction and deep sequencing. Nat. Methods 2013, 10, 133–139. [Google Scholar] [CrossRef]
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 1991, 252, 1651–1656. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef]
- Brown, K.M.; Gilmartin, G.M. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol. Cell 2003, 12, 1467–1476. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Camarillo, C.; Orozco, E.; Marchat, L.A. Entamoeba histolytica: Comparative genomics of the pre-mRNA 3′ end processing machinery. Exp. Parasitol. 2005, 110, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lutz, C.S.; Wilusz, J.; Tian, B. Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 2005, 11, 1485–1493. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, C.J.; Schmidt, R.; Kanitz, A.; Artimo, P.; Gruber, A.J.; Zavolan, M. PolyASite 2.0: A consolidated atlas of polyadenylation sites from 3’ end sequencing. Nucleic Acids Res. 2020, 48, D174–D179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Nambiar, R.; Zheng, D.; Tian, B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018, 46, D315–D319. [Google Scholar] [CrossRef]
- Müller, S.; Rycak, L.; Afonso-Grunz, F.; Winter, P.; Zawada, A.M.; Damrath, E.; Scheider, J.; Schmäh, J.; Koch, I.; Kahl, G.; et al. APADB: A database for alternative polyadenylation and microRNA regulation events. Database 2014, 2014, bau076. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wu, J.; Feng, Y.; Fu, Y.; Guo, Y.; Long, L.; Zhang, H.; Luan, Y.; Tian, P.; Chen, L.; et al. APASdb: A database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res. 2015, 43, D59–D67. [Google Scholar] [CrossRef]
- Kostadinov, R.; Malhotra, N.; Viotti, M.; Shine, R.; D’Antonio, L.; Bagga, P. GRSDB: A database of quadruplex forming G-rich sequences in alternatively processed mammalian pre-mRNA sequences. Nucleic Acids Res. 2006, 34, D119–D124. [Google Scholar] [CrossRef] [Green Version]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Contreras-Moreira, B.; Castro-Mondragon, J.A.; Santana-Garcia, W.; Ossio, R.; Robles-Espinoza, C.D.; Bahin, M.; Collombet, S.; Vincens, P.; Thieffry, D.; et al. RSAT 2018: Regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 2018, 46, W209–W214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Mathews, D.H.; Turner, D.H.; Watson, R.M. RNA secondary structure prediction. Curr. Protoc. Nucleic Acid. Chem. 2016, 67, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.R.; Bernhart, S.H.; Lorenz, R. The ViennaRNA web services. Methods Mol. Biol. 2015, 1269, 307–326. [Google Scholar] [CrossRef]
- Akhtar, M.N.; Bukhari, S.A.; Fazal, Z.; Qamar, R.; Shahmuradov, I.A. POLYAR, a new computer program for prediction of poly(A) sites in human sequences. BMC Genomics. 2010, 11, 646. [Google Scholar] [CrossRef] [Green Version]
- Ji, G.; Wu, X.; Shen, Y.; Huang, J.; Quinn Li, Q. A classification-based prediction model of messenger RNA polyadenylation sites. J. Theor. Biol. 2010, 265, 287–296. [Google Scholar] [CrossRef]
- Tzanis, G.; Kavakiotis, I.; Vlahavas, I. PolyA-iEP: A data mining method for the effective prediction of polyadenylation sites. Expert Syst Appl. 2011, 38, 12398–12408. [Google Scholar] [CrossRef]
- Cheng, Y.; Miura, R.M.; Tian, B. Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics 2006, 22, 2320–2325. [Google Scholar] [CrossRef] [Green Version]
- Magana-Mora, A.; Kalkatawi, M.; Bajic, V.B. Omni-PolyA: A method and tool for accurate recognition of Poly(A) signals in human genomic DNA. BMC Genom. 2017, 18, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Tian, B. APAlyzer: A bioinformatics package for analysis of alternative polyadenylation isoforms. Bioinformatics 2020, 36, 3907–3909. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.J. Hidden Markov models and their applications in biological sequence analysis. Curr. Genom. 2009, 10, 402–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouchka, E.C.; Wang, X.; Graham, J.H.; Cooper, N.G.F. Computational prediction of genes translationally regulated by cytoplasmic polyadenylation elements. In Bioinformatics and Computational Biology; Rajasekaran, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 5462, pp. 353–361. [Google Scholar] [CrossRef]
- Van Helden, J. Regulatory sequence analysis tools. Nucleic Acids Res. 2003, 31, 3593–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.; Gruber, A.R.; Keller, W.; Zavolan, M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012, 1, 753–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soemedi, R.; Cygan, K.J.; Rhine, C.L.; Glidden, D.T.; Taggart, A.J.; Lin, C.L.; Fredericks, A.M.; Fairbrother, W.G. The effects of structure on pre-mRNA processing and stability. Methods 2017, 125, 36–44. [Google Scholar] [CrossRef]
- Kwok, C.K. Dawn of the in vivo RNA structurome and interactome. Biochem. Soc. Trans. 2016, 44, 1395–1410. [Google Scholar] [CrossRef]
- Lu, Z.; Chang, H.Y. Decoding the RNA structurome. Curr. Opin. Struct. Biol. 2016, 36, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Kubota, M.; Chan, D.; Spitale, R.C. RNA structure: Merging chemistry and genomics for a holistic perspective. Bioessays 2015, 37, 1129–1138. [Google Scholar] [CrossRef]
- Kwok, C.K.; Tang, Y.; Assmann, S.M.; Bevilacqua, P.C. The RNA structurome: Transcriptome-wide structure probing with next-generation sequencing. Trends Biochem. Sci. 2015, 40, 221–232. [Google Scholar] [CrossRef]
- Bernat, V.; Disney, M.D. RNA Structures as mediators of neurological diseases and as drug targets. Neuron 2015, 87, 28–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevilacqua, P.C.; Ritchey, L.E.; Su, Z.; Assmann, S.M. Genome-wide analysis of RNA secondary structure. Annu. Rev. Genet. 2016, 50, 235–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, S.E.; Pyle, A.M. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc. Chem. Res. 2011, 44, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Hurst, T.; Xu, X.; Zhao, P.; Chen, S.J. Quantitative understanding of SHAPE mechanism from RNA structure and dynamics analysis. J. Phys. Chem. B 2018, 122, 4771–4783. [Google Scholar] [CrossRef]
- Mitchell, D., 3rd; Assmann, S.M.; Bevilacqua, P.C. Probing RNA structure in vivo. Curr. Opin. Struct. Biol. 2019, 59, 151–158. [Google Scholar] [CrossRef]
- Aviran, S.; Pachter, L. Rational experiment design for sequencing-based RNA structure mapping. RNA 2014, 20, 1864–1877. [Google Scholar] [CrossRef] [Green Version]
- Puton, T.; Kozlowski, L.P.; Rother, K.M.; Bujnicki, J.M. CompaRNA: A server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 2014, 42, 5403–5406. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Do, C.B.; Woods, D.A.; Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006, 22, e90–e98. [Google Scholar] [CrossRef]
- Sato, K.; Hamada, M.; Asai, K.; Mituyama, T. Centroidfold: A web server for RNA secondary structure prediction. Nucleic Acids Res. 2009, 37, W277–W280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Kato, Y.; Hamada, M.; Akutsu, T.; Asai, K. IPknot: Fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011, 27, i85–i93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Shira, A.; Panet, A.; Honigman, A. An RNA secondary structure juxtaposes two remote genetic signals for human T-cell leukemia virus type I RNA 3′-end processing. J. Virol. 1991, 65, 5165–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graveley, B.R.; Fleming, E.S.; Gilmartin, G.M. RNA structure is a critical determinant of poly(A) site recognition by cleavage and polyadenylation specificity factor. Mol. Cell Biol. 1996, 16, 4942–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klasens, B.I.; Das, A.T.; Berkhout, B. Inhibition of polyadenylation by stable RNA secondary structure. Nucleic Acids Res. 1998, 26, 1870–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klasens, B.I.; Thiesen, M.; Virtanen, A.; Berkhout, B. The ability of the HIV-1 AAUAAA signal to bind polyadenylation factors is controlled by local RNA structure. Nucleic Acids Res. 1999, 27, 446–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passarelli, A.L.; Guarino, L.A. Baculovirus late and very late gene regulation. Curr Drug Targets. 2007, 8, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- van Oers, M.M.; Vlak, J.M.; Voorma, H.O.; Thomas, A.A.M. Role of the 3′ untranslated region of baculovirus p10 mRNA in high-level expression of foreign genes. J. Gen. Virol. 1999, 80, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zhong, S.; Fei, Z.; Hashimoto, Y.; Xiang, J.Z.; Zhang, S.; Blissard, G.W. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 2013, 87, 6391–6405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Balázs, Z.; Kis, E.; Molnár, J.; Boldogkői, Z. Third-generation sequencing reveals extensive polycistronism and transcriptional overlapping in a baculovirus. Sci. Rep. 2018, 8, 8604. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Guarino, L.A. 3’-end formation of baculovirus late RNAs. J. Virol. 2000, 74, 8930–8937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.Q.; Chen, K.P.; Yao, Q.; Guo, Z.J.; Wang, L.L. Characterization of a late gene, ORF67 from Bombyx mori nucleopolyhedrovirus. FEBS Lett. 2007, 581, 5836–5842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, A.; Gopinathan, K.P. Characterization of late gene expression factors lef-9 and lef-8 from Bombyx mori nucleopolyhedrovirus. J. Gen. Virol. 2002, 83, 2015–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.F.; Yin, X.M.; Guo, Z.J.; Zhu, L.J. Characterization of a late gene, ORF60 from Bombyx mori nucleopolyhedrovirus. J. Biochem. Mol. Biol. 2006, 39, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Guarino, L.A. Processing of baculovirus late and very late mRNAs. Virol. Sin. 2007, 22, 108–116. [Google Scholar] [CrossRef]
- Pilloff, M.G.; Bilen, M.F.; Belaich, M.N.; Lozano, M.E.; Ghiringhelli, P.D. Molecular cloning and sequence analysis of the Anticarsia gemmatalis multicapsid nuclear polyhedrosis virus GP64 glycoprotein. Virus Genes 2003, 26, 57–69. [Google Scholar] [CrossRef]
- Passarelli, A.L.; Todd, J.W.; Miller, L.K. A baculovirus gene involved in late gene expression predicts a large polypeptide with a conserved motif of RNA polymerases. J. Virol. 1994, 68, 4673–4678. [Google Scholar] [CrossRef] [Green Version]
- Crouch, E.A.; Cox, L.T.; Morales, K.G.; Passarelli, A.L. Inter-subunit interactions of the Autographa californica M nucleopolyhedrovirus RNA polymerase. Virology 2007, 367, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Ruprich-Robert, G.; Thuriaux, P. Non-canonical DNA transcription enzymes and the conservation of two-barrel RNA polymerases. Nucleic Acids Res. 2010, 38, 4559–4569. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioinformatic Tool | Description | URL | Reference | |
|---|---|---|---|---|
| Databases | EST databases | contain sets of short cDNA sequences (500–800 nt) representing fragments of expressed genes from wide-diverse transcriptomes; used for transcripts identification and gene sequence determination | https://www.ncbi.nlm.nih.gov/genbank/dbest/ | [61] |
| PolyASite | portal to curated sets of human, mouse, and worm poly(A) sites, based on all 3′ end sequencing datasets available in the SRA nucleotide database (June 2019) | https://polyasite.unibas.ch/ | [68] | |
| PolyA_DB3 | contains poly(A) sites identified in several vertebrate species | https://exon.apps.wistar.org/PolyA_DB/ | [69] | |
| APADB | database for mammalian APA determined by 3′ end sequencing | http://tools.genxpro.net/apadb/ | [70] | |
| APASdb | database of APA sites designed to visualize the precise map and usage quantification of different APA isoforms on a genome-wide scale for all genes | http://genome.bucm.edu.cn/utr/ | [71] | |
| GRSDB - The ‘G’-Rich Sequences Database | contains information on composition and distribution of putative quadruplex forming ‘G’-Rich Sequences (QGRS) in the alternatively processed (alternatively spliced or alternatively polyadenylated) mammalian pre-mRNA sequences | https://bioinformatics.ramapo.edu/grsdb/index.php | [72] | |
| Alignments | Clustal | software for multiple sequence alignment which algorithm proceeds in a three-steps-routine, including pairwise alignment, distance matrix determination and guide tree creation to align the query sequences depending on their similarity | http://www.clustal.org/ | [64] |
| Muscle | software for MSA which algorithm is based on a three-stages-routine, consisting in a draft multiple alignment creation, its re-estimation using the Kimura distance algorithm producing a superior draft alignment, and a final refinement stage | http://www.drive5.com/muscle/ | [65] | |
| HMMER | fast and sensitive homology searches using profile hidden Markov Models | http://hmmer.org/https://www.ebi.ac.uk/Tools/hmmer/ | [73] | |
| Pattern search | MEME SUITE | online server for sequence motifs discovery and analysis | http://meme-suite.org/ | [74] |
| RSAT | analysis tools for cis-regulatory elements in genome sequences | http://rsat.sb-roscoff.fr/ | [75] | |
| Sequence Logos | graphical representation of the sequence conservation in biological sequences (DNA, RNA, and proteins) created from an MSA | https://weblogo.berkeley.edu/http://weblogo.threeplusone.com/ | [76] | |
| Structure determination | Mfold | web server for nucleic acid folding and hybridization prediction | http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form | [77] |
| RNAstructure | web server for RNA secondary structure prediction | http://rna.urmc.rochester.edu/RNAstructureWeb/ | [78] | |
| ViennaRNA Web Services | provide programs, web services and databases related to RNA secondary structures | http://rna.tbi.univie.ac.at/ | [79] | |
| PASes prediction | POLYAR | software for PASes prediction in human sequences, based on PAS and CS functional characterization and their distance determination | http://www.mybiosoftware.com/polyar-human-polyadenylation-site-prediction.html | [80] |
| PAC | recognition model for PASes prediction in plant sequences with a modular design and adaptable to other species | http://www.polya.org/ | [81] | |
| PolyA-iEP | data mining method for PASes prediction in A. Thaliana, determining emerging patterns and used for descriptive and predictive analysis | http://mlkd.csd.auth.gr/PolyA/index.html | [82] | |
| PolyA_SVM | program for poly(A) sites prediction in DNA/RNA sequences and/or determines the occurrence of cis-elements | https://exon.apps.wistar.org/polya_svm/ | [83] | |
| Omni-PolyA | recognition model for human PASes prediction, based on the combination of machine learning and genetic algorithms | https://www.cbrc.kaust.edu.sa/omnipolya/ | [84] | |
| APAlyzer | performs 3′-UTR APA, intronic APA and gene expression analysis using RNA-Seq data | https://bioconductor.org/packages/release/bioc/html/APAlyzer.html | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peros, I.G.; Cerrudo, C.S.; Pilloff, M.G.; Belaich, M.N.; Lozano, M.E.; Ghiringhelli, P.D. Advances in the Bioinformatics Knowledge of mRNA Polyadenylation in Baculovirus Genes. Viruses 2020, 12, 1395. https://doi.org/10.3390/v12121395
Peros IG, Cerrudo CS, Pilloff MG, Belaich MN, Lozano ME, Ghiringhelli PD. Advances in the Bioinformatics Knowledge of mRNA Polyadenylation in Baculovirus Genes. Viruses. 2020; 12(12):1395. https://doi.org/10.3390/v12121395
Chicago/Turabian StylePeros, Iván Gabriel, Carolina Susana Cerrudo, Marcela Gabriela Pilloff, Mariano Nicolás Belaich, Mario Enrique Lozano, and Pablo Daniel Ghiringhelli. 2020. "Advances in the Bioinformatics Knowledge of mRNA Polyadenylation in Baculovirus Genes" Viruses 12, no. 12: 1395. https://doi.org/10.3390/v12121395
APA StylePeros, I. G., Cerrudo, C. S., Pilloff, M. G., Belaich, M. N., Lozano, M. E., & Ghiringhelli, P. D. (2020). Advances in the Bioinformatics Knowledge of mRNA Polyadenylation in Baculovirus Genes. Viruses, 12(12), 1395. https://doi.org/10.3390/v12121395