
Binding of CCCTC-Binding Factor (CTCF) to the Minute Virus of Mice Genome Is Important for Proper Processing of Viral P4-Generated Pre-mRNAs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Viruses
2.2. Transfections and Viral Infections
2.3. Cell Synchronization
2.4. Plasmids
2.5. Extraction of MVMp Nucleoprotein Complexes
2.6. Total RNA Isolation
2.7. RNase Protection (RPA) Assay
2.8. Northern Blotting
2.9. Chromatin Immunoprecipitation (ChIP) Assay in Whole Cell Lysates
2.10. Chromatin Immunoprecipitation (ChIP) Assay on Viral Nucleoprotein Complexes
2.11. Immunoblot Analysis
2.12. Southern Blot Analysis
2.13. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and TA Cloning
2.14. Immunofluorescence Assay
3. Results
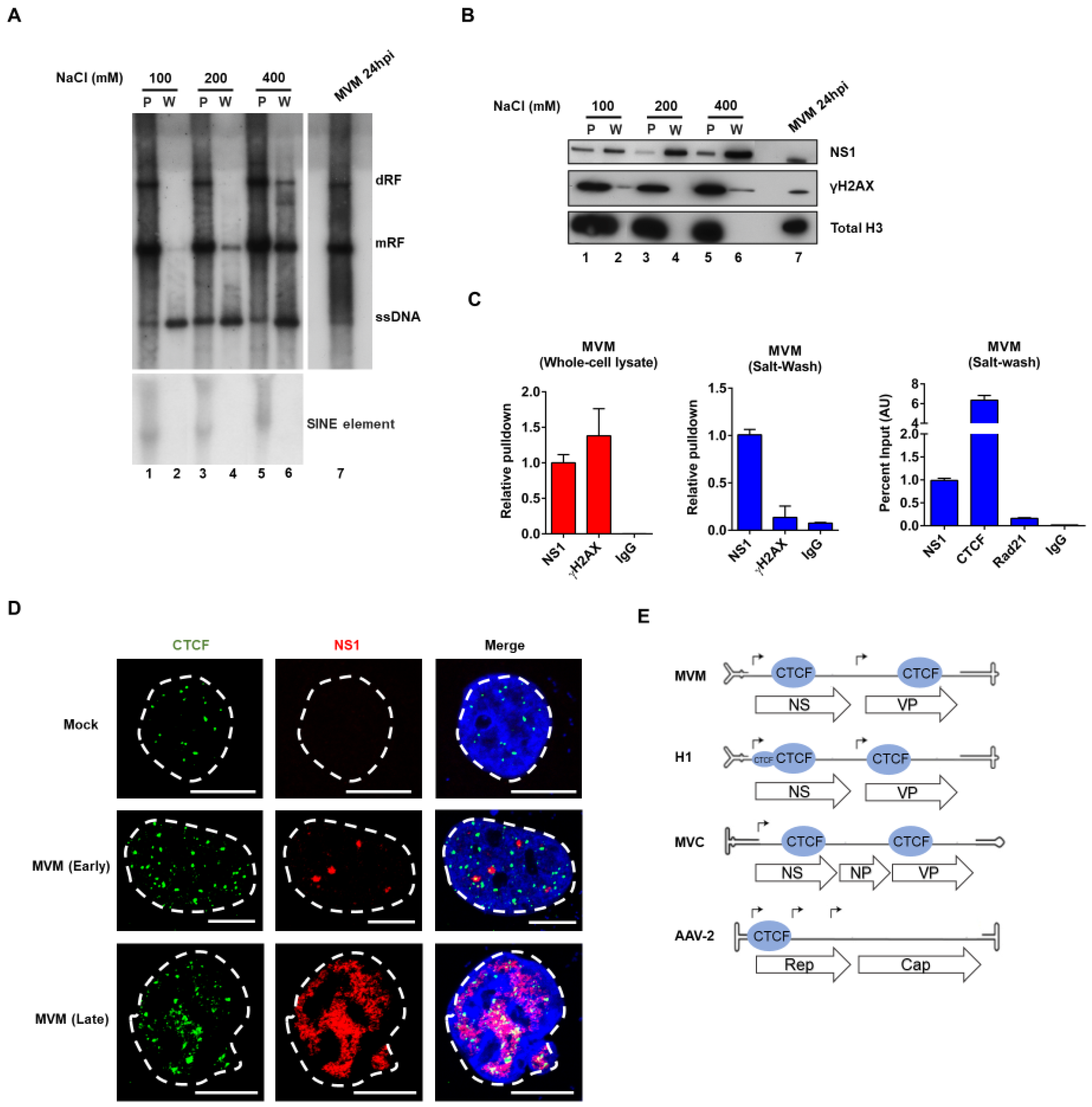
3.1. CTCF Specifically Binds the Viral Genome and Localizes to MVM Replication Compartments
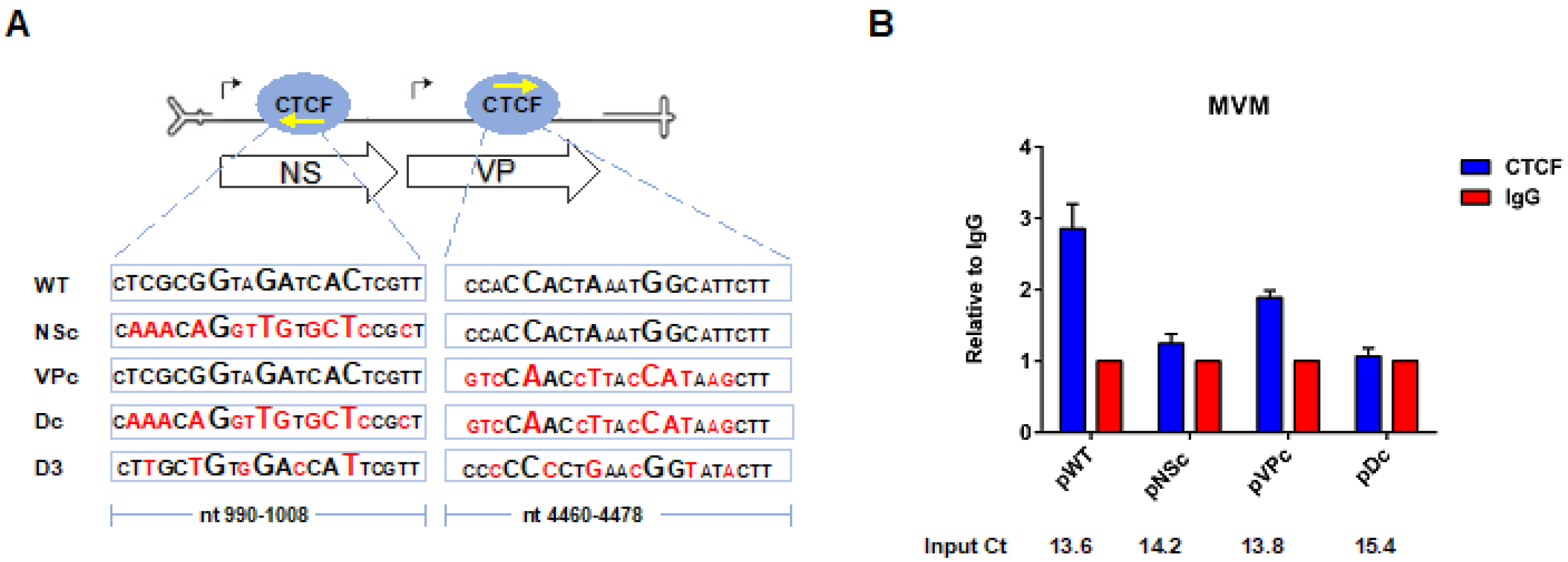
3.2. CTCF-Binding Site Mutants Exhibited a Decrease in Levels of Spliced to Unspliced R1, as Well as Reduced Levels of R2 Relative to R1
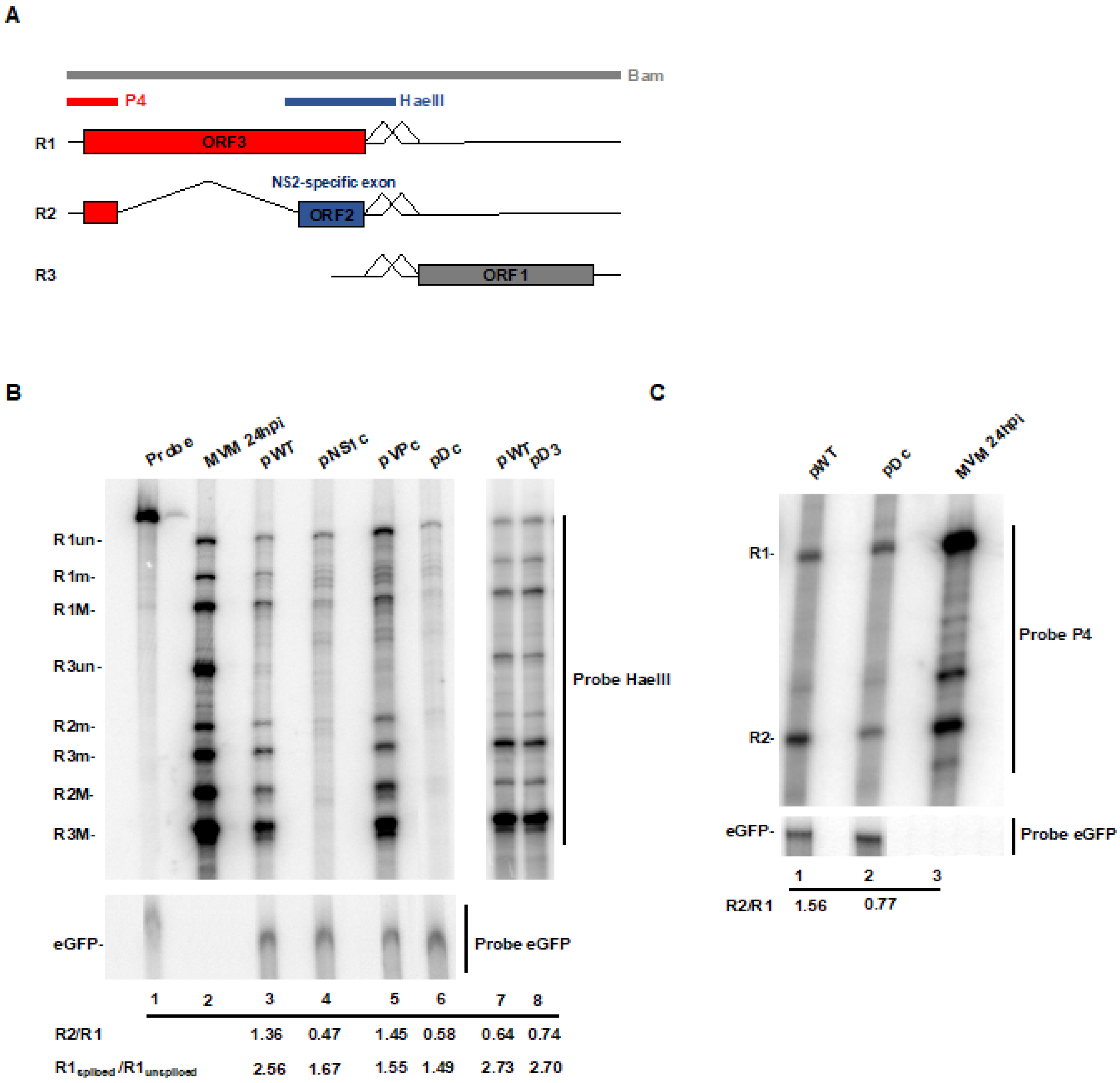
3.3. CTCF-Binding Site Mutants Resulted in Skipping of the NS2-Specific Exon and Joining of the Large Intron Donor to the Small Intron Acceptors
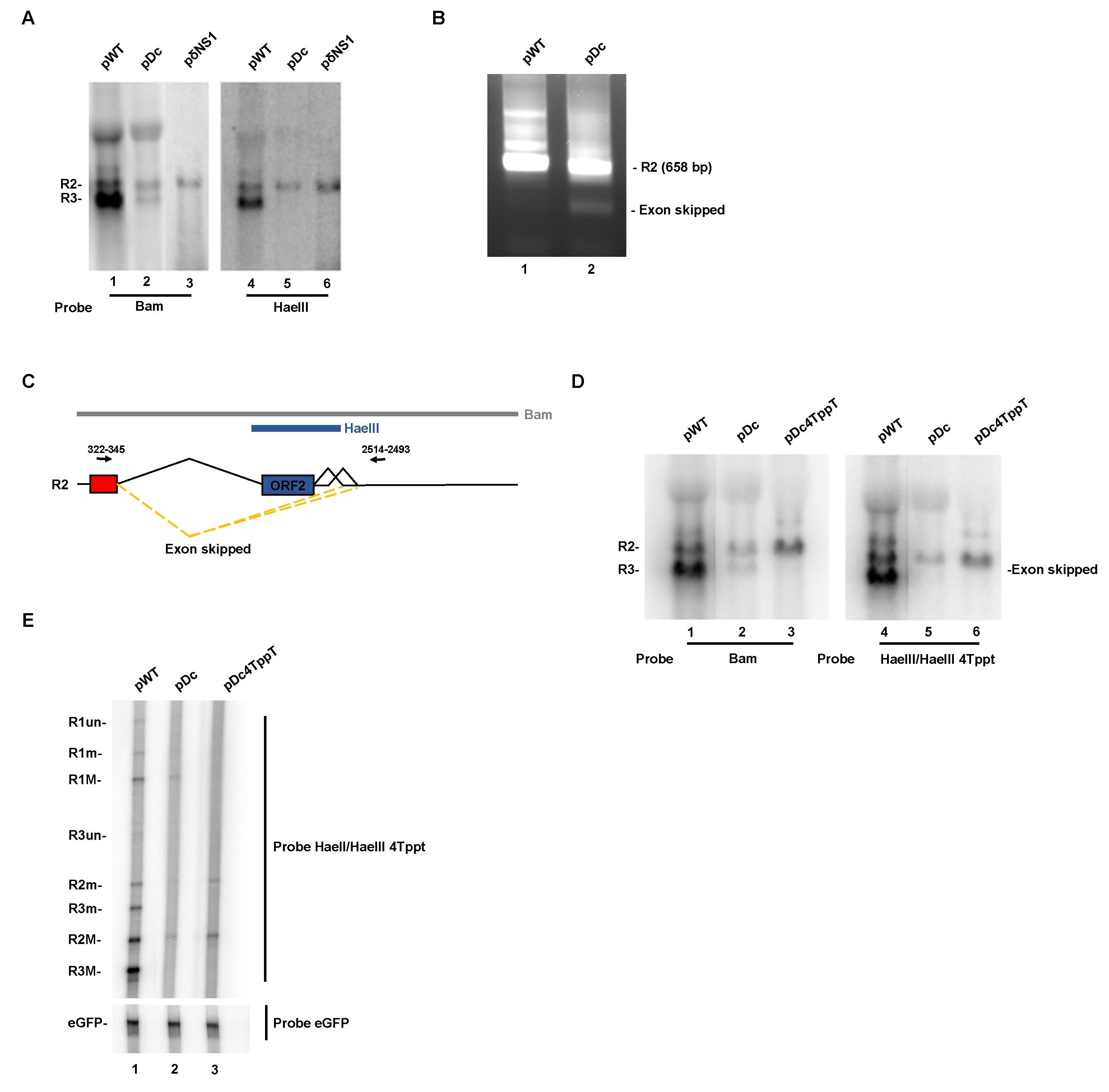
3.4. Improvement of the Large Intron Splice Acceptor in the Dc Mutant Led to Increased NS2-Specific Exon Definition and Increased Levels of R2 RNA
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintel, D.; Dadachanji, D.; Astell, C.R.; Ward, D.C. The genome of minute virus of mice, an autonomous parvovirus, encodes two overlapping transcription units. Nucleic Acids Res. 1983, 11, 1019–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotmore, S.F.; Tattersall, P. Organization of nonstructural genes of the autonomous parvovirus minute virus of mice. J. Virol. 1986, 58, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labieniec-Pintel, L.; Pintel, D. The minute virus of mice P39 transcription unit can encode both capsid proteins. J. Virol. 1986, 57, 1163–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemens, K.E.; Cerutis, D.R.; Burger, L.R.; Yang, C.Q.; Pintel, D.J. Cloning of minute virus of mice cDNAs and preliminary analysis of individual viral proteins expressed in murine cells. J. Virol. 1990, 64, 3967–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongeneel, C.V.; Sahli, R.; McMaster, G.K.; Hirt, B. A precise map of splice junctions in the mRNAs of minute virus of mice, an autonomous parvovirus. J. Virol. 1986, 59, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Morgan, W.R.; Ward, D.C. Three splicing patterns are used to excise the small intron common to all minute virus of mice RNAs. J. Virol. 1986, 60, 1170–1174. [Google Scholar] [CrossRef] [Green Version]
- Schoborg, R.V.; Pintel, D.J. Accumulation of MVM gene products is differentially regulated by transcription initiation, RNA processing and protein stability. Virology 1991, 181, 22–34. [Google Scholar] [CrossRef]
- Pintel, D.J.; Gersappe, A.; Haut, D.; Pearson, J. Determinants that govern alternative splicing of parvovirus pre-mRNAs. Semin. Virol. 1995, 6, 283–290. [Google Scholar] [CrossRef]
- Bashir, T.; Rommelaere, J.; Cziepluch, C. In Vivo Accumulation of Cyclin A and Cellular Replication Factors in Autonomous Parvovirus Minute Virus of Mice-Associated Replication Bodies. J. Virol. 2001, 75, 4394–4398. [Google Scholar] [CrossRef] [Green Version]
- Adeyemi, R.O.; Landry, S.; Davis, M.E.; Weitzman, M.D.; Pintel, D.J. Parvovirus minute virus of mice induces a DNA damage response that facilitates viral replication. PLoS Pathog. 2010, 6, e1001141. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, Z.; Mihaylov, I.S.; Cotmore, S.F.; Tattersall, P. Recruitment of DNA replication and damage response proteins to viral replication centers during infection with NS2 mutants of Minute Virus of Mice (MVM). Virology 2011, 410, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, K.; Wang, J.; Boftsi, M.; Fuller, M.S.; Rede, J.E.; Joshi, T.; Pintel, D.J. Parvovirus minute virus of mice interacts with sites of cellular DNA damage to establish and amplify its lytic infection. Elife 2018, 7. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Phillips-Cremins, J.E.; Sauria, M.E.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.; Ong, C.T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013, 153, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.; Ye, Z.; Kolovos, P.; Brouwer, R.W.; van de Corput, M.P.; van de Werken, H.J.; Knoch, T.A.; van, I.W.F.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [Green Version]
- Braccioli, L.; de Wit, E. CTCF: A Swiss-army knife for genome organization and transcription regulation. Essays Biochem. 2019, 63, 157–165. [Google Scholar] [CrossRef]
- Saldana-Meyer, R.; Gonzalez-Buendia, E.; Guerrero, G.; Narendra, V.; Bonasio, R.; Recillas-Targa, F.; Reinberg, D. CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 2014, 28, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldana-Meyer, R.; Rodriguez-Hernaez, J.; Escobar, T.; Nishana, M.; Jacome-Lopez, K.; Nora, E.P.; Bruneau, B.G.; Tsirigos, A.; Furlan-Magaril, M.; Skok, J.; et al. RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol. Cell 2019, 76, 412–422.e5. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Velasco, M.; Kumar, M.; Lai, M.C.; Bhat, P.; Solis-Pinson, A.B.; Reyes, A.; Kleinsorg, S.; Noh, K.M.; Gibson, T.J.; Zaugg, J.B. CTCF-Mediated Chromatin Loops between Promoter and Gene Body Regulate Alternative Splicing across Individuals. Cell Syst. 2017, 5, 628–637.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Lieberman, P.M. Cell Cycle Control of Kaposi’s Sarcoma-Associated Herpesvirus Latency Transcription by CTCF-Cohesin Interactions. J. Virol. 2009, 83, 6199–6210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, C.M.; Zhang, X.-Y.; McMahon, S.B.; Lieberman, P.M. Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J. Virol. 2006, 80, 5723–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, F.P.; Cruz, R.; Lu, F.; Plasschaert, R.; Deng, Z.; Rivera-Molina, Y.A.; Bartolomei, M.S.; Lieberman, P.M.; Tang, Q. CTCF binding to the first intron of the major immediate early (MIE) gene of human cytomegalovirus (HCMV) negatively regulates MIE gene expression and HCMV replication. J. Virol. 2014, 88, 7389–7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.-J.; Verma, D.; Mosbruger, T.; Swaminathan, S. CTCF and Rad21 act as host cell restriction factors for Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog. 2014, 10, e1003880. [Google Scholar] [CrossRef]
- Paris, C.; Pentland, I.; Groves, I.; Roberts, D.C.; Powis, S.J.; Coleman, N.; Roberts, S.; Parish, J.L. CCCTC-binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J. Virol. 2015, 89, 4770–4785. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Schoborg, R.V.; Pintel, D.J. Alternative splicing of pre-mRNAs encoding the nonstructural proteins of minute virus of mice is facilitated by sequences within the downstream intron. J. Virol. 1994, 68, 2849–2859. [Google Scholar] [CrossRef] [Green Version]
- Naeger, L.K.; Cater, J.; Pintel, D.J. The small nonstructural protein (NS2) of the parvovirus minute virus of mice is required for efficient DNA replication and infectious virus production in a cell-type-specific manner. J. Virol. 1990, 64, 6166–6175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerig, C.; McMaster, G.; Sogo, J.; Bruggmann, H.; Beard, P. Nucleoprotein complexes of minute virus of mice have a distinct structure different from that of chromatin. J. Virol. 1986, 58, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blissenbach, M.; Grewe, B.; Hoffmann, B.; Brandt, S.; Überla, K. Nuclear RNA Export and Packaging Functions of HIV-1 Rev Revisited. J. Virol. 2010, 84, 6598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, L.K.; Fasina, O.; Pintel, D.J. RNAse Mapping and Quantitation of RNA Isoforms. In RNA Abundance Analysis: Methods and Protocols; Jin, H., Gassmann, W., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 121–129. [Google Scholar]
- Fuller, M.S.; Majumder, K.; Pintel, D.J. Minute Virus of Mice Inhibits Transcription of the Cyclin B1 Gene during Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Nasmyth, K.; Haering, C.H. Cohesin: Its Roles and Mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Gersappe, A.; Pintel, D.J. Efficient excision of the upstream large intron from P4-generated pre-mRNA of the parvovirus minute virus of mice requires at least one donor and the 3′ splice site of the small downstream intron. J. Virol. 1995, 69, 6170–6179. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Mathur, S.; Burger, L.R.; Pintel, D.J. Sequences within the parvovirus minute virus of mice NS2-specific exon are required for inclusion of this exon into spliced steady-state RNA. J. Virol. 1995, 69, 5864–5868. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Fasina, O.O.; Pintel, D.J. Minute Virus of Canines NP1 Protein Interacts with the Cellular Factor CPSF6 To Regulate Viral Alternative RNA Processing. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Hsin, J.P.; Manley, J.L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Pintel, D.J. The adeno-associated virus type 2 Rep protein regulates RNA processing via interaction with the transcription template. Mol. Cell. Biol. 2002, 22, 3639–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, K.D.; Pintel, D.J. Improved splicing of adeno-associated viral (AAV) capsid protein-supplying pre-mRNAs leads to increased recombinant AAV vector production. Hum. Gene Ther. 2008, 19, 1421–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.-Y.; Newman, A.E.; Burger, L.; Pintel, D. Replication of minute virus of mice DNA is critically dependent on accumulated levels of NS2. J. Virol. 2005, 79, 12375–12381. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boftsi, M.; Majumder, K.; Burger, L.R.; Pintel, D.J. Binding of CCCTC-Binding Factor (CTCF) to the Minute Virus of Mice Genome Is Important for Proper Processing of Viral P4-Generated Pre-mRNAs. Viruses 2020, 12, 1368. https://doi.org/10.3390/v12121368
Boftsi M, Majumder K, Burger LR, Pintel DJ. Binding of CCCTC-Binding Factor (CTCF) to the Minute Virus of Mice Genome Is Important for Proper Processing of Viral P4-Generated Pre-mRNAs. Viruses. 2020; 12(12):1368. https://doi.org/10.3390/v12121368
Chicago/Turabian StyleBoftsi, Maria, Kinjal Majumder, Lisa R. Burger, and David J. Pintel. 2020. "Binding of CCCTC-Binding Factor (CTCF) to the Minute Virus of Mice Genome Is Important for Proper Processing of Viral P4-Generated Pre-mRNAs" Viruses 12, no. 12: 1368. https://doi.org/10.3390/v12121368
APA StyleBoftsi, M., Majumder, K., Burger, L. R., & Pintel, D. J. (2020). Binding of CCCTC-Binding Factor (CTCF) to the Minute Virus of Mice Genome Is Important for Proper Processing of Viral P4-Generated Pre-mRNAs. Viruses, 12(12), 1368. https://doi.org/10.3390/v12121368