
Modeling the Molecular Impact of SARS-CoV-2 Infection on the Renin-Angiotensin System



Abstract
1. Introduction
2. Methods
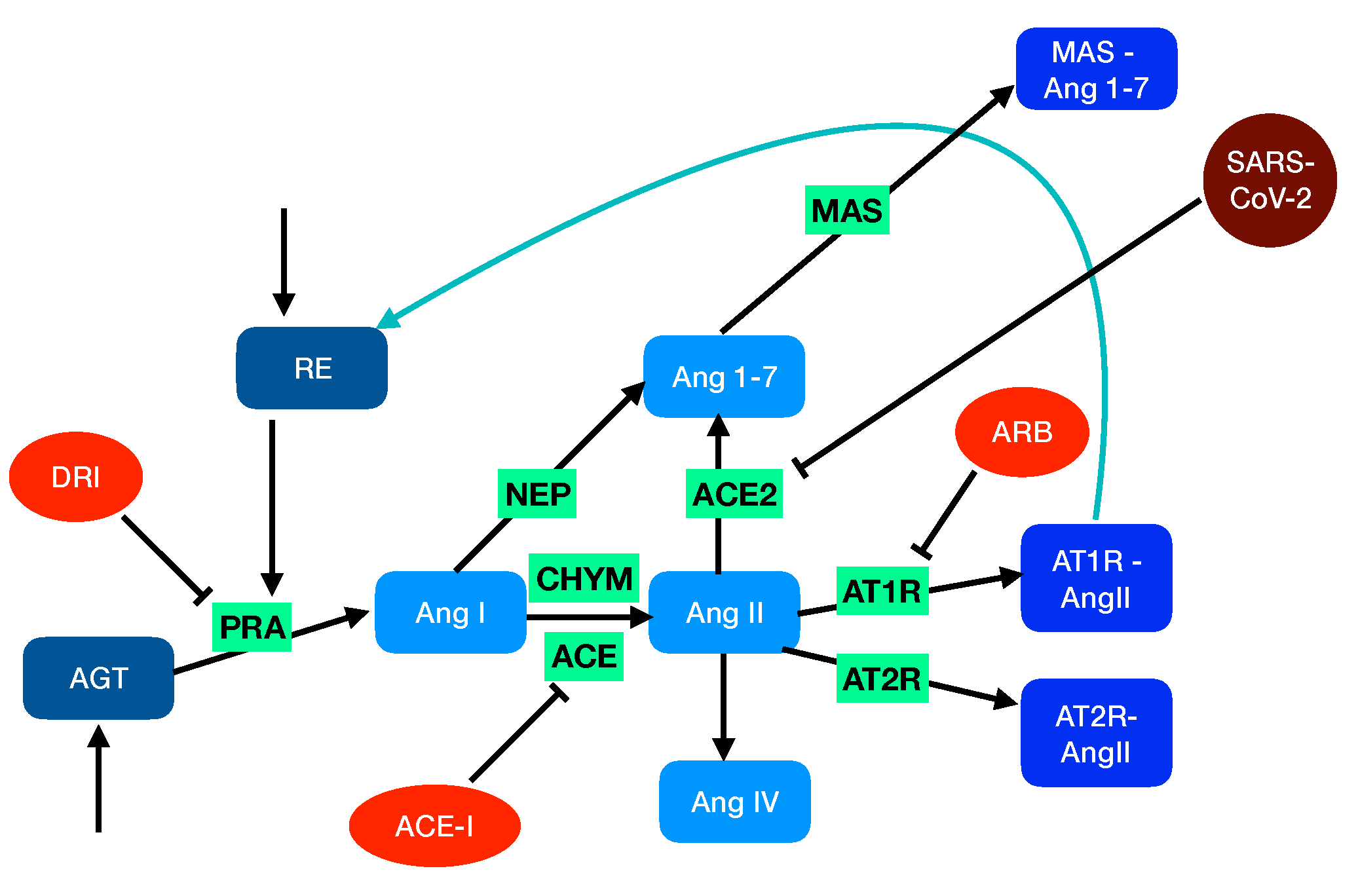
2.1. Modeling the Renin-Angiotensin System
- The angiotensin-converting enzyme (ACE, EC3.4.15.1) is a zinc metalloproteinase located mainly in the capillaries of lung and in the endothelial cells. It catalyzes the transformation of AngI into the octapeptide angiotensin II (AngII).
- Chymase (CHY, EC 3.4.21.39), a serine protease that is mainly localized in blood vessels and heart, also catalyzes the transformation of AngI into AngII.
- Neprilysin (NEP, EC3.4.24.11), another zinc metalloproteinase that is expressed in a wide variety of tissues, catalyzes the transformation of AngI into the heptapeptide hormone angiotensin-(1-7) (Ang1-7).
2.2. Modeling Blood Pressure
2.3. Modeling RAS-Blocker Effects
- Angiotensin-converting enzyme inhibitors (ACE-I) that bind to ACE and thus inhibit the formation of angiotensin II and the associated vasoconstriction and inflammatory cascades. Examples of this type of drug are enalapril, lisinopril, and captopril.
- Angiotensin receptor blockers (ARB) that block the binding of AngII to AT1R and thus act in antagonism with AngII. Examples are candesartan, losartan, and valsartan.
- Direct renin inhibitors (DRI) that act on renin and thus inhibit the conversion of AGT to AngI. Examples are aliskiren, enalkiren, and remikiren.
2.4. Modeling SARS-CoV-2 Infection
2.5. Modeling ARDS Severity
2.6. Solving the RAS Model
2.7. Stability of the RAS Model
3. Results
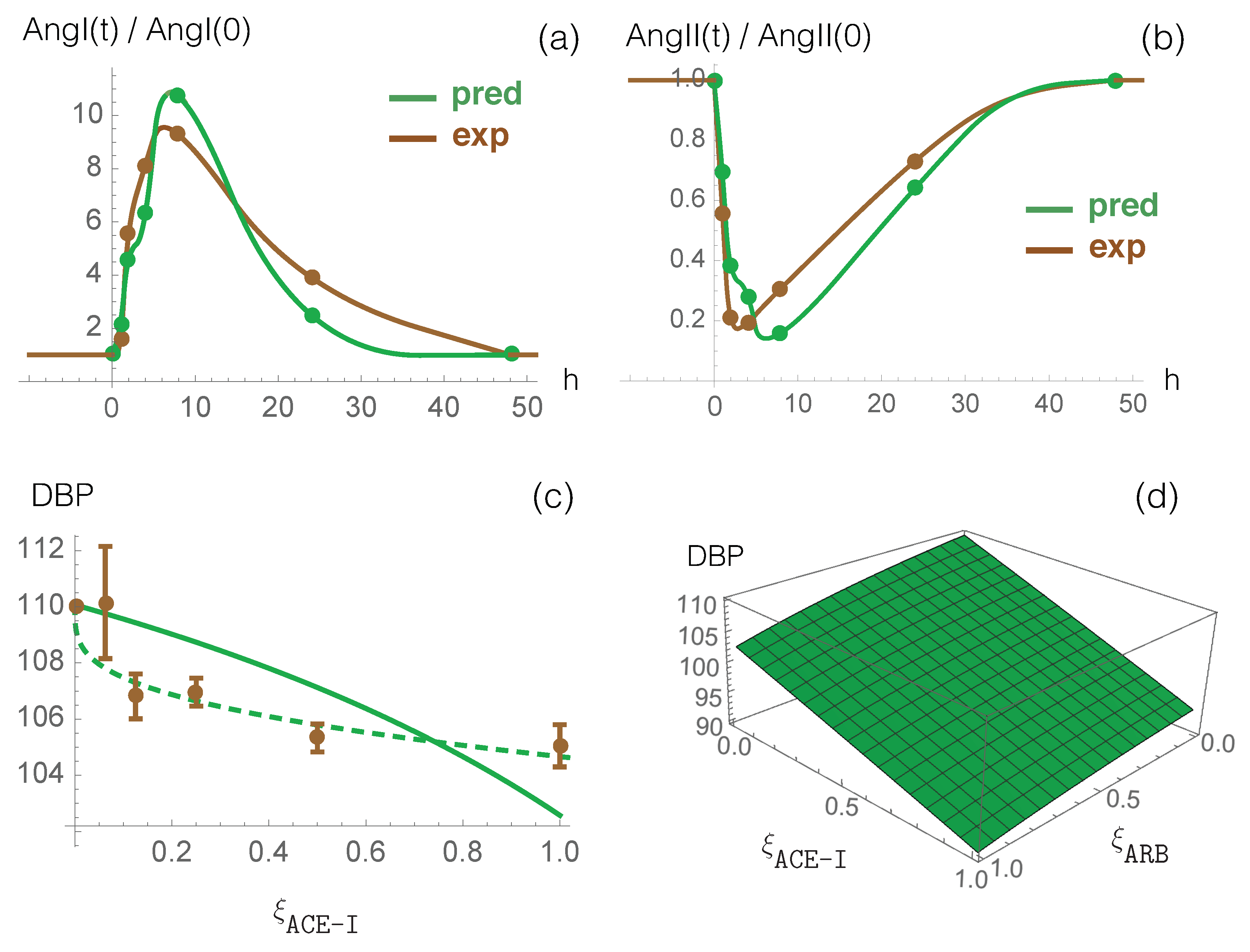
3.1. Model Predictions and Clinical Data on RAS-Blocker Drugs
3.2. RAS in COVID-19
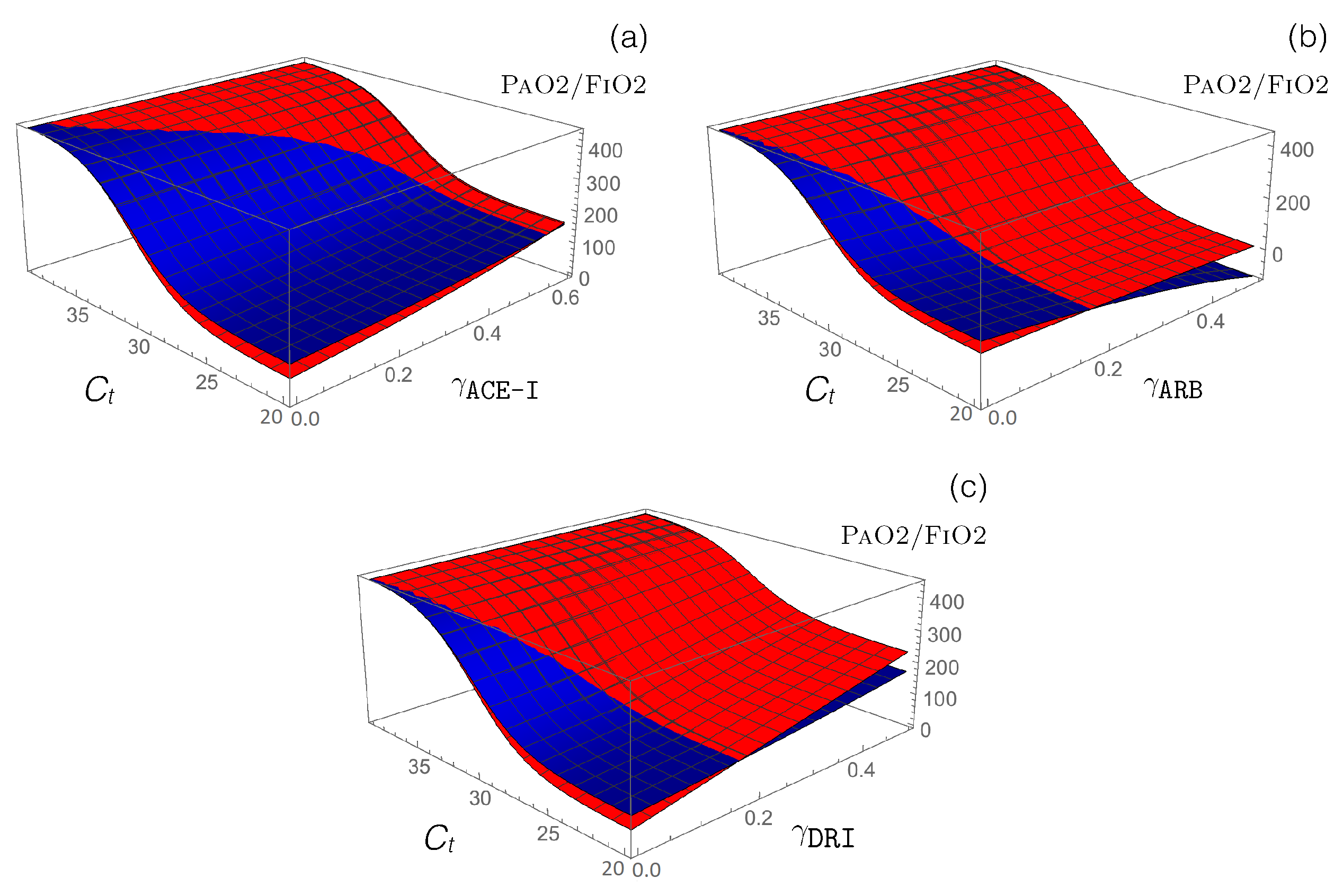
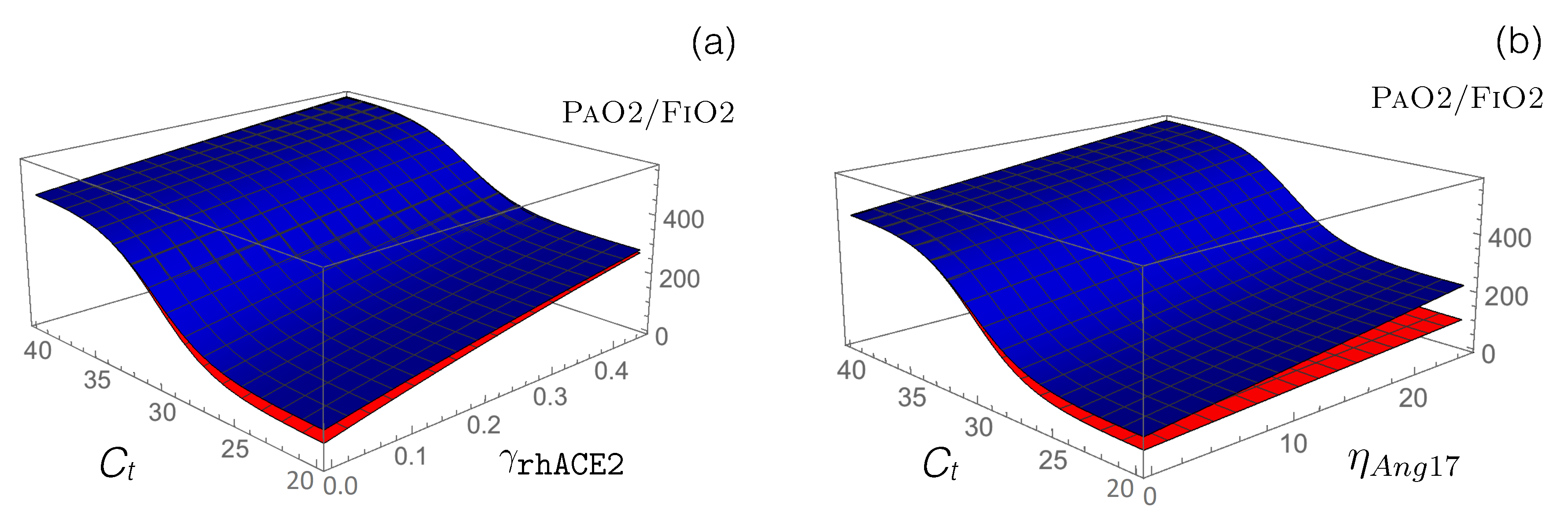
3.3. Impact of RAS-Modulating Drugs on COVID-19 Severity
- Antihypertensive RAS-blocking drugs: We combined the effect of each of the three RAS-blocking ACE-I, ARB, and DRI drugs, which were modeled by the enzyme-inhibiting functions (introduced in Equation (12)), with the ACE2-inhibiting -dependent function (defined in Equation (14)), which mimics SARS-CoV-2 infection. the PaO2/FiO2 values predicted by our model are presented in Figure 4.
- Other RAS-targeting drugs: We used our model to test the potential of other drugs that are currently in clinical trials to restore the functional activity of the perturbed RAS upon viral infection. First, we modeled how the administration of an exogenous supplement of rhACE2 (GSK2586881) affects RAS by modifying the reaction rate defined in Equation (13). This rate already includes the function that mimics SARS-CoV-2 infection, and we simply added a second function associated with the effects of rhACE2 administration:
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Yuan, S.; Kok, K.H.; To, K.K.W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.Y.; Poon, R.W.S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Benvenuto, D.; Giovanetti, M.; Ciccozzi, A.; Spoto, S.; Angeletti, S.; Ciccozzi, M. The 2019-new coronavirus epidemic: Evidence for virus evolution. J. Med. Virol. 2020, 92, 455–459. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020, 30, 1346–1351.e2. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin–angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin- converting enzyme cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- NCT04287686. Recombinant Human Angiotensin-Converting Enzyme 2 (rhACE2) as a Treatment for Patients with COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04287686 (accessed on 27 February 2020).
- NCT04332666. Angiotensin-(1,7) Treatment in COVID-19: The ATCO Trial (ATCO). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04332666 (accessed on 3 April 2020).
- NCT04335786. Valsartan for Prevention of Acute Respiratory Distress Syndrome in Hospitalized Patients With SARS-COV-2 (COVID-19) Infection Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT04335786 (accessed on 6 April 2020).
- NCT04312009. Losartan for Patients with COVID-19 Requiring Hospitalization. Available online: https://clinicaltrials.gov/ct2/show/NCT04312009 (accessed on 17 March 2020).
- NCT04311177. Losartan for Patients with COVID-19 Not Requiring Hospitalization. Available online: https://clinicaltrials.gov/ct2/show/NCT04311177 (accessed on 17 March 2020).
- NCT04318418. ACE Inhibitors, Angiotensin II Type-I Receptor Blockers and Severity of COVID-19 (CODIV-ACE). Available online: https://clinicaltrials.gov/ct2/show/NCT04318418 (accessed on 24 March 2020).
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin–angiotensin–aldosterone system inhibitors and risk of Covid-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin–angiotensin–aldosterone system blockers and the risk of Covid-19. N. Engl. J. Med. 2020, 382, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Mehra, M.R.; Desai, S.S.; Kuy, S.; Henry, T.D.; Patel, A.N. Cardiovascular disease, drug therapy, and mortality in COVID-19. N. Engl. J. Med. 2020, 382, e102. [Google Scholar] [CrossRef]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Raizada, M.K.; Phillips, M.I.; Sumners, C. Cellular and Molecular Biology of the Renin-Angiotensin System; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Casarini, D.E.; Arita, D.Y.; Cunha, T.S.; Colucci, J.A. New Aspects of the Renin Angiotensin System in Cardiovascular and Renal Diseases; Bentham: Sharjah, UAE, 2016. [Google Scholar]
- Hallow, K.M.; Lo, A.; Beh, J.; Rodrigo, M.; Ermakov, S.; Friedman, S.; de Leon, H.; Sarkar, A.; Xiong, Y.; Sarangapani, R.; et al. A model-based approach to investigating the pathophysiological mechanisms of hypertension and response to antihypertensive therapies: Extending the Guyton model. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R647–R662. [Google Scholar] [CrossRef]
- Versypt, A.N.F.; Harrell, G.K.; McPeak, A.N. A pharmacokinetic/pharmacodynamic model of ACE inhibition of the renin-angiotensin system for normal and impaired renal function. Comput. Chem. Eng. 2017, 104, 311–322. [Google Scholar] [CrossRef]
- Leete, J.; Gurley, S.; Layton, A.T. Modeling sex differences in the renin angiotensin system and the efficacy of antihypertensive therapies. Comput. Chem. Eng. 2018, 112, 253–264. [Google Scholar] [CrossRef]
- Leete, J.; Layton, A.T. Sex-specific long-term blood pressure regulation: Modeling and analysis. Comput. Biol. Med. 2019, 104, 139–148. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Suzuki, Y.; Mezzano, S.; Plaza, J.; Egido, J. Role of the renin-angiotensin system in vascular diseases: Expanding the field. Hypertension 2001, 38, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- De Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; François, C.; Schalij, I.; Dorfmüller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated renin–angiotensin–aldosterone system contributes to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension 2018, 72, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.J. Hypertension: Renin–angiotensin– aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The intrarenal renin-angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef]
- Simões e Silva, A.; Silveira, K.; Ferreira, A.; Teixeira, M. ACE2, angiotensin-(1-7) and M as receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef]
- Povlsen, A.L.; Grimm, D.; Wehland, M.; Infanger, M.; Krüger, M. The vasoactive Mas receptor in essential hypertension. J. Clin. Med. 2020, 9, 267. [Google Scholar] [CrossRef]
- Santos, R.A.; e Silva, A.C.S.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef]
- Carey, R.M. AT2 receptors: Potential therapeutic targets for hypertension. Am. J. Hypertens. 2017, 30, 339–347. [Google Scholar]
- Zaman, M.A.; Oparil, S.; Calhoun, D.A. Drugs targeting the renin–angiotensin–aldosterone system. Nat. Rev. Drug Discov. 2002, 1, 621–636. [Google Scholar] [CrossRef]
- Williams, B. Drug discovery in renin–angiotensin system intervention: Past and future. Ther. Adv. Cardiovasc. Dis. 2016, 10, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Borg, I.; Rohde, G.; Löseke, S.; Bittscheidt, J.; Schultze-Werninghaus, G.; Stephan, V.; Bufe, A. Evaluation of a quantitative real-time PCR for the detection of respiratory syncytial virus in pulmonary diseases. Eur. Respir. J. 2003, 21, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Fan, J.; Yu, F.; Feng, B.; Lou, B.; Zou, Q.; Xie, G.; Lin, S.; Wang, R.; Yang, X.; et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: Retrospective cohort study. BMJ 2020, 369, m1443. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Pérez-Méndez, L.; Blanco, J.; Añón, J.M.; Blanch, L.; Belda, J.; Santos-Bouza, A.; Fernández, R.L.; Kacmarek, R.M. A universal definition of ARDS: The PaO2/FiO2 ratio under a standard ventilatory setting—a prospective, multicenter validation study. Intensive Care Med. 2013, 39, 583–592. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. New Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Streatfeild-James, R.M.; Williamson, D.; Pike, R.N.; Tewksbury, D.; Carrell, R.W.; Coughlin, P.B. Angiotensinogen cleavage by renin: Importance of a structurally constrained N-terminus. FEBS Lett. 1998, 436, 267–270. [Google Scholar] [CrossRef]
- Katsurada, A.; Hagiwara, Y.; Miyashita, K.; Satou, R.; Miyata, K.; Ohashi, N.; Navar, L.G.; Kobori, H. Novel sandwich ELISA for human angiotensinogen. Am. J. Physiol. Ren. Physiol. 2007, 293, F956–F960. [Google Scholar] [CrossRef]
- Chappell, M.C. Biochemical evaluation of the renin-angiotensin system: The good, bad, and absolute? Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H137–H152. [Google Scholar] [CrossRef]
- Pendergrass, K.D.; Pirro, N.T.; Westwood, B.M.; Ferrario, C.M.; Brosnihan, K.B.; Chappell, M.C. Sex differences in circulating and renal angiotensins of hypertensive mRen. Lewis but not normotensive Lewis rats. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H10–H20. [Google Scholar] [CrossRef]
- Sullivan, J.C.; Rodriguez-Miguelez, P.; Zimmerman, M.A.; Harris, R.A. Differences in angiotensin (1–7) between men and women. Am. J. Physiol.-Heart Circ. Physiol. 2015, 308, H1171–H1176. [Google Scholar] [CrossRef]
- Nussberger, J.; Brunner, D.B.; Waeber, B.; Brunner, H.R. Specific measurement of angiotensin metabolites and in vitro generated angiotensin II in plasma. Hypertension 1986, 8, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Nussberger, J.; Brunner, D.; Keller, I.; Brunner, H.R. Measurement of converting enzyme activity by antibody-trapping of generated angiotensin II: Comparison with two other methods. Am. J. Hypertens. 1992, 5, 393–398. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mazzolai, L.; Maillard, M.; Rossat, J.; Nussberger, J.; Brunner, H.R.; Burnier, M. Angiotensin II receptor blockade in normotensive subjects: A direct comparison of three AT1 receptor antagonists. Hypertension 1999, 33, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Nussberger, J.; Wuerzner, G.; Jensen, C.; Brunner, H.R. Angiotensin II Suppression in humans by the orally active renin inhibitor aliskiren (SPP100) comparison with enalapril. Hypertension 2002, 39, e1–e8. [Google Scholar] [CrossRef]
- Heran, B.S.; Wong, M.M.; Heran, I.K.; Wright, J.M. Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Doulton, T.W.; He, F.J.; MacGregor, G.A. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005, 45, 880–886. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 1–5. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Liu, N.; Hong, Y.; Chen, R.G.; Zhu, H.M. High rate of increased level of plasma Angiotensin II and its gender difference in COVID-19: An analysis of 55 hospitalized patients with COVID-19 in a single hospital, WuHan, China. medRxiv 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr. Hypertens. Rep. 2018, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E.; Lazos, S.A.; Tong, K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J. Clin. Investig. 1995, 95, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef]
- Ssentongo, A.; Ssentongo, P.; Heilbrunn, E.S.; Lekoubou, A.; Du, P.; Liao, D.; Oh, J.S.; Chinchilli, V.M. Renin-angiotensin-aldosterone system inhibitors and mortality in patients with hypertension hospitalized for COVID-19: A systematic review and meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Khera, R.; Clark, C.; Lu, Y.; Guo, Y.; Ren, S.; Truax, B.; Spatz, E.S.; Murugiah, K.; Lin, Z.; Omer, S.B.; et al. Association of Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers with the Risk of Hospitalization and Death in Hypertensive Patients with Coronavirus Disease-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Baral, R.; White, M.; Vassiliou, V.S. Impact of hospitalised patients with COVID-19 taking Renin-Angiotensin-Aldosterone System inhibitors: A systematic review and meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Zambelli, V.; Bellani, G.; Borsa, R.; Pozzi, F.; Grassi, A.; Scanziani, M.; Castiglioni, V.; Masson, S.; Decio, A.; Laffey, J.G.; et al. Angiotensin-(1-7) improves oxygenation, while reducing cellular infiltrate and fibrosis in experimental Acute Respiratory Distress Syndrome. Intensive Care Med. Exp. 2015, 3, 1–17. [Google Scholar] [CrossRef]
- Chester, A.; Borland, J. Chymase-dependent angiotensin II formation in human blood vessels. J. Hum. Hypertens. 2000, 14, 373–376. [Google Scholar] [CrossRef]
- Athyros, V.G.; Mikhailidis, D.P.; Kakafika, A.I.; Tziomalos, K.; Karagiannis, A. Angiotensin II reactivation and aldosterone escape phenomena in renin–angiotensin–aldosterone system blockade: Is oral renin inhibition the solution? Expert Opin. Pharmacother. 2007, 8, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Amaya, F.; Oh-hashi, K.; Kiuchi, K.; Hashimoto, S. Expression of neutral endopeptidase activity during clinical and experimental acute lung injury. Respir. Res. 2010, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Morant-Talamante, N.; Lupón, J. Neprilysin and natriuretic peptide regulation in heart failure. Curr. Heart Fail. Rep. 2016, 13, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Acanfora, D.; Ciccone, M.M.; Scicchitano, P.; Acanfora, C.; Casucci, G. Neprilysin inhibitor–angiotensin II receptor blocker combination (sacubitril/valsartan): Rationale for adoption in SARS-CoV-2 patients. Eur. Heart-J.–Cardiovasc. Pharmacother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Crowley, S.D.; Rudemiller, N.P. Immunologic effects of the renin-angiotensin system. J. Am. Soc. Nephrol. 2017, 28, 1350–1361. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Values | Reference |
|---|---|---|---|
| min | 600 | [35] | |
| min | 0.5 | [35] | |
| min | 0.5 | [35] | |
| min | 0.5 | [35] | |
| min | 0.5 | [35] | |
| min | 12 | [35] | |
| min | 12 | [35] | |
| min | 12 | [35] | |
| min | 12 | - | |
| 1/min | 20 | [36,54] | |
| mmHg | 450 | Fitted | |
| mmHg | 267 | Fitted | |
| mmHg | 73.6 | Fitted | |
| mmHg mL/fmol | 0.43 | Fitted | |
| a | - | 0.53 | Fitted |
| b | - | 16.7 | Fitted |
| Parameter | Unit | Normotensive | Hypertensive | Reference |
|---|---|---|---|---|
| [AGT] | fmol/mL | 6 | 6 | [55] |
| [AngI] | fmol/mL | 70 | 110 | [56,57] |
| [AngII] | fmol/mL | 28 | 156 | [56,57] |
| [Ang1-7] | fmol/mL | 36 | 92 | [56,57,58] |
| [AngIV] | fmol/mL | 1 | 1 | [59] |
| [AT1R-AngII] | fmol/mL | 15 | 85 | [37] |
| [AT2R-AngII] | fmol/mL | 5 | 27 | [37] |
| [RE] | fmol/mL | 9.43 | 25.25 | Solved |
| [MAS-Ang1-7] | fmol/mL | 6.43 | 15.92 | Solved |
| fmol/(mL min) | 881.82 | 1198.22 | Solved | |
| fmol/(mL min) | 0.54 | 2.21 | Solved | |
| 1/min | 1.31 | 3.21 | Solved | |
| 1/min | 1.80 | 0.82 | Solved | |
| 1/min | 0.05 | 0.01 | Solved | |
| 1/min | 0.03 | 0.03 | Solved | |
| 1/min | 0.01 | 0.01 | Solved |
| Drugs | Class | Dose | [AngI](t)/[AngI] | [AngII](t)/[AngII] | Np | Ref. |
|---|---|---|---|---|---|---|
| (mg) | rmsd (Range) | rmsd (Range) | ||||
| Enalapril | ACE-I | 20 | 1.31 [1.0–9.2] | 0.09 [0.2–1.0] | 5 | [60] |
| Losartan | ARB | 50 | 0.61 [1.0–2.1] | - | 3 | [61] |
| Valsartan | ARB | 850 | 0.83 [1.0–2.2] | - | 3 | [61] |
| Irbesartan | ARB | 150 | 0.97 [1.0–4.4] | - | 3 | [61] |
| Aliskiren | DRI | 40 | 0.13 [0.4–1.1] | 0.14 [0.5–1.0] | 6 | [62] |
| Aliskiren | DRI | 80 | 0.15 [0.4–1.0] | 0.16 [0.4–1.0] | 6 | [62] |
| Aliskiren | DRI | 160 | 0.26 [0.2–1.0] | 0.20 [0.3–1.0] | 6 | [62] |
| Aliskiren | DRI | 640 | 0.29 [0.1–1.0] | 0.29 [0.1–1.0] | 6 | [62] |
| Mean | 0.57 | 0.18 | ||||
| Uninfected | Mild | Moderate | Severe | |
|---|---|---|---|---|
| 40.0 | 31.5 | 27.6 | 23.8 | |
| Normotensive | ||||
| [AngII] (fmol/mL) | 28 | 32 | 36 | 38 |
| [Ang1-7] (fmol/mL) | 36 | 21 | 5 | 1 |
| PaO2/FiO2 (mmHg) | 450 | 300 | 145 | 98 |
| DBP (mmHg) | 80 | 81 | 82 | 82 |
| Hypertensive | ||||
| [AngII] (fmol/mL) | 156 | 186 | 221 | 231 |
| [Ang1-7] (fmol/mL) | 92 | 55 | 15 | 2 |
| PaO2/FiO2 (mmHg) | 450 | 292 | 115 | 60 |
| DBP (mmHg) | 110 | 117 | 125 | 128 |
| Drugs | No Drugs | ACE-I | ARB | DRI | rhACE2 | Ang1–7 |
|---|---|---|---|---|---|---|
| Normotensive—Moderate Infection | ||||||
| [AngII]/[AngII] | 1.29 | 1.10 | 1.98 | 0.99 | 1.10 | 1.29 |
| [Ang1-7]/[Ang1-7] | 0.15 | 0.13 | 0.23 | 0.11 | 0.68 | 0.64 |
| PaO2/FiO2 (mmHg) | 145 | 188 | 0 | 216 | 337 | 278 |
| DBP (mmHg) | 82 | 81 | 80 | 80 | 81 | 82 |
| Hypertensive—Moderate Infection | ||||||
| [AngII]/[AngII] | 1.42 | 1.12 | 1.55 | 0.77 | 1.14 | 1.42 |
| [Ang1-7]/[Ang1-7] | 0.16 | 0.13 | 0.18 | 0.09 | 0.70 | 0.36 |
| PaO2/FiO2 (mmHg) | 115 | 185 | 83 | 268 | 332 | 167 |
| DBP (mmHg) | 125 | 114 | 101 | 102 | 115 | 125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pucci, F.; Bogaerts, P.; Rooman, M. Modeling the Molecular Impact of SARS-CoV-2 Infection on the Renin-Angiotensin System. Viruses 2020, 12, 1367. https://doi.org/10.3390/v12121367
Pucci F, Bogaerts P, Rooman M. Modeling the Molecular Impact of SARS-CoV-2 Infection on the Renin-Angiotensin System. Viruses. 2020; 12(12):1367. https://doi.org/10.3390/v12121367
Chicago/Turabian StylePucci, Fabrizio, Philippe Bogaerts, and Marianne Rooman. 2020. "Modeling the Molecular Impact of SARS-CoV-2 Infection on the Renin-Angiotensin System" Viruses 12, no. 12: 1367. https://doi.org/10.3390/v12121367
APA StylePucci, F., Bogaerts, P., & Rooman, M. (2020). Modeling the Molecular Impact of SARS-CoV-2 Infection on the Renin-Angiotensin System. Viruses, 12(12), 1367. https://doi.org/10.3390/v12121367