Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus


Abstract

1. Gene Therapy Using Viral Vectors
2. Lentiviruses and Gene Therapy
3. Molecular Biology of Lentiviruses
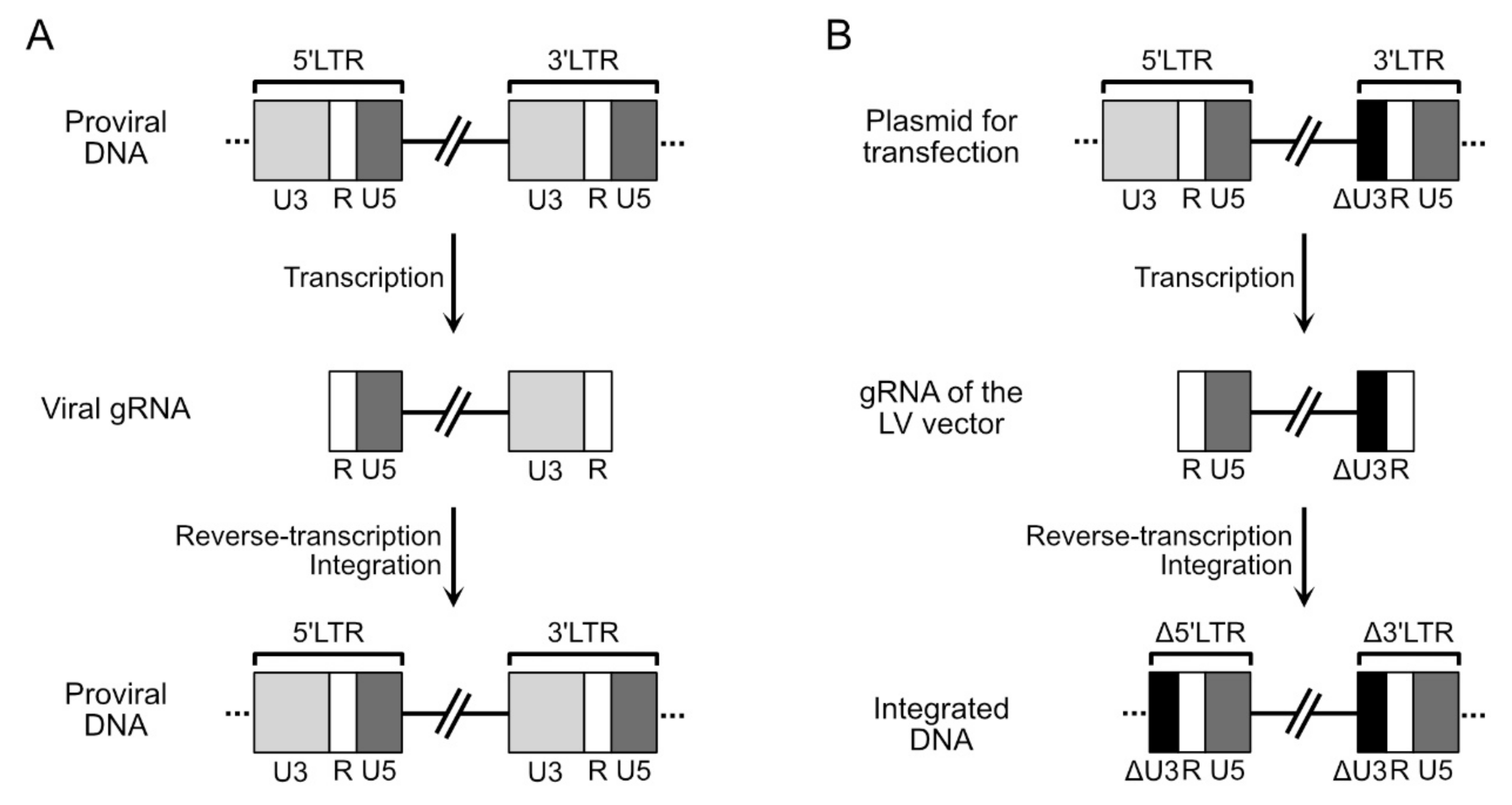
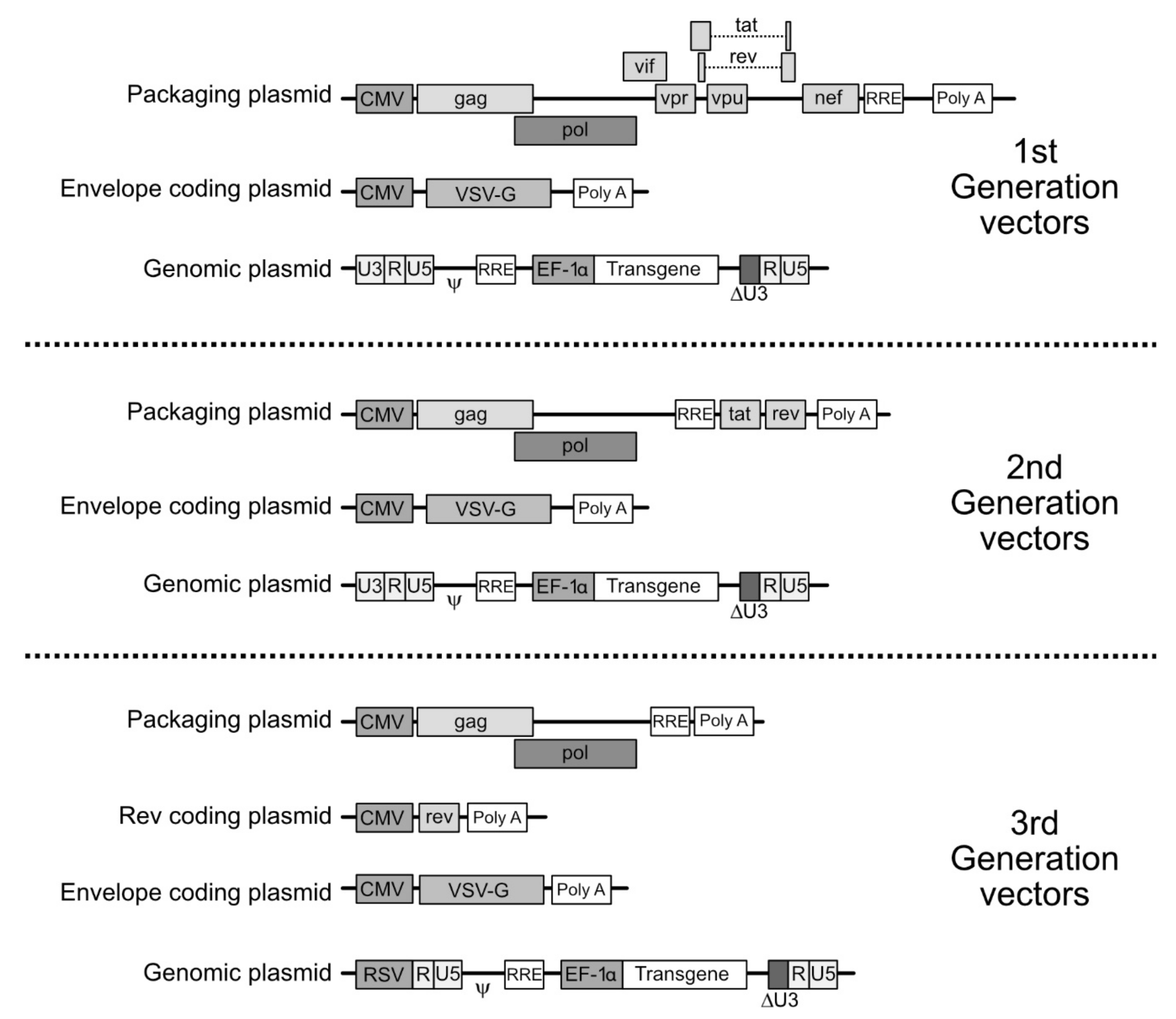
4. Molecular Bases for the Making of LV Vectors
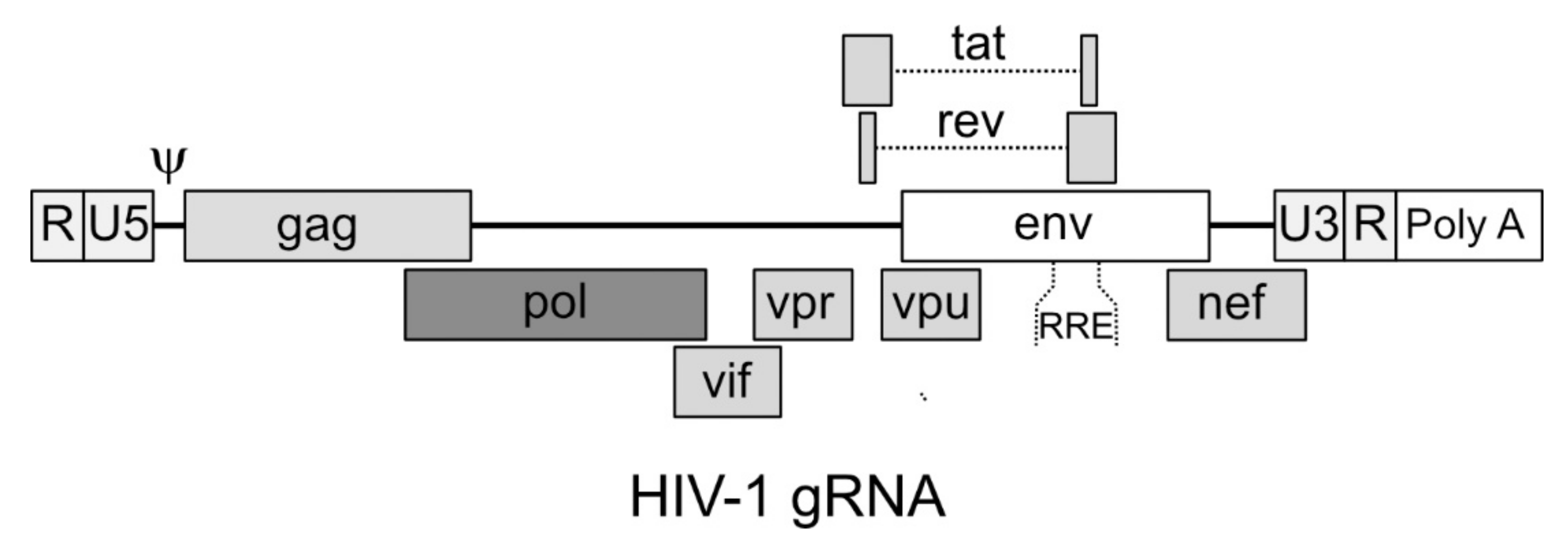
4.1. Structure of the Genomic RNA
4.2. Mechanism of Entry in HIV-1
4.3. Mechanism of Recruitment of HIV-1 Envelope Proteins on the Surface of the Virus: Bases for Pseudotyping
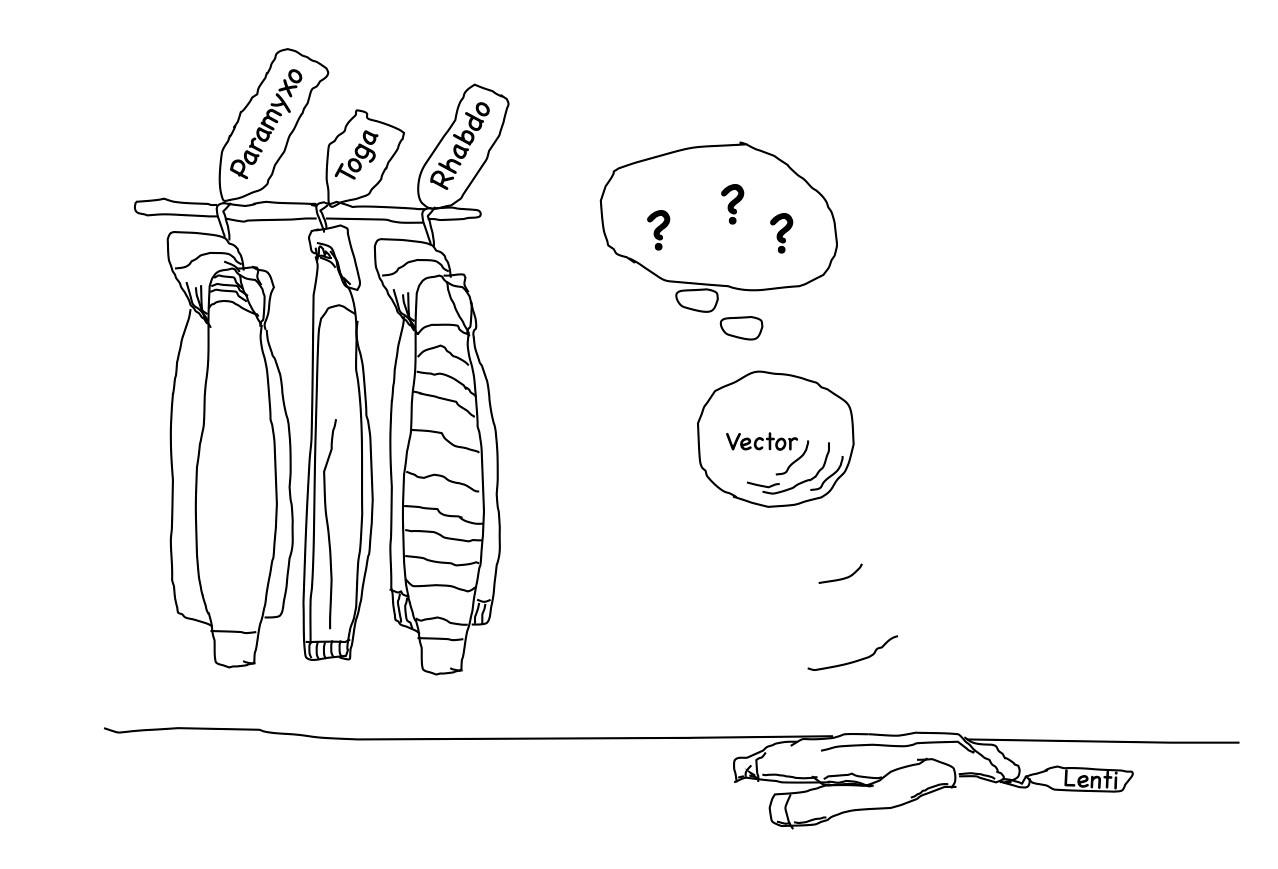
5. Dressing LV Vectors (Pseudotyping)
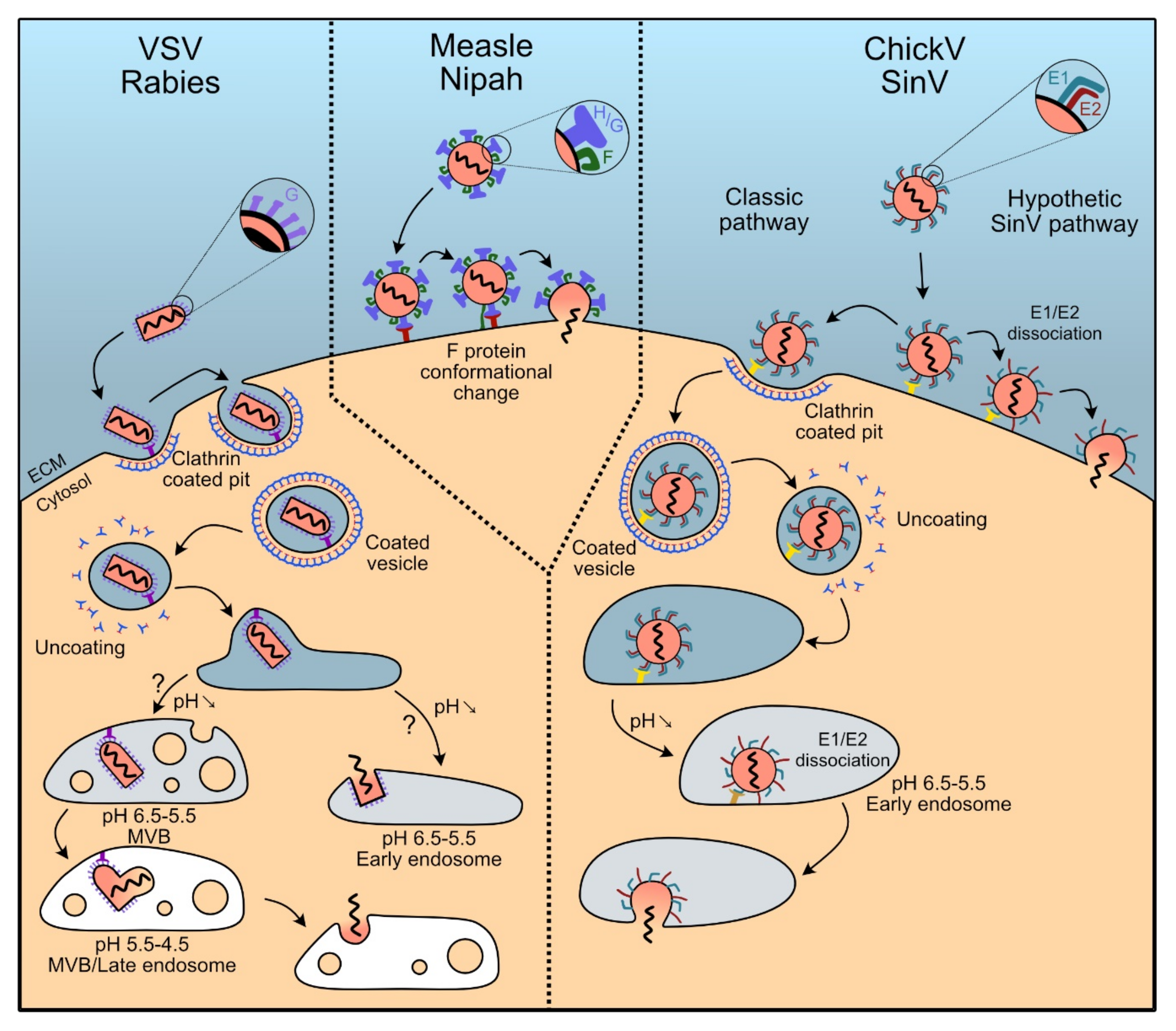
5.1. Rhabdoviruses: Clathrin-Dependent Endocytosis
5.1.1. Vescicular Stomatitis Virus
5.1.2. Rabies Virus
5.2. Paramyxoviruses: Splitting Binding and Fusion in H and F Proteins
5.2.1. Measles Virus
5.2.2. Nipah Virus
5.3. Togaviridae: The Pair E1-E2 Dissociates to Trigger Membrane Fusion
5.3.1. Chikungunya Virus
5.3.2. Sindbis Virus
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- U.S. National Library of Medicine. What is Gene Therapy? Genetics Home Reference—NIH. Available online: https://ghr.nlm.nih.gov/primer/therapy/genetherapy (accessed on 27 August 2020).
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.B.; Büning, H.; Galy, A.; Schambach, A.; Grez, M. Gene therapy on the move. EMBO Mol. Med. 2013, 5, 1642–1661. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Zonari, E.; DeSantis, G.; Petrillo, C.; Boccalatte, F.E.; Lidonnici, M.R.; Kajaste-Rudnitski, A.; Aiuti, A.; Ferrari, G.; Naldini, L.; Gentner, B. Efficient Ex Vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep. 2017, 8, 977–990. [Google Scholar] [CrossRef]
- Gowing, G.; Svendsen, S.; Svendsen, C.N. Ex Vivo gene therapy for the treatment of neurological disorders. In Progress in Brain Research; Elsevier BV: Amsterdam, The Netherlands, 2017; Volume 230, pp. 99–132. [Google Scholar]
- Rosenberg, S.A. Gene Therapy for Cancer. JAMA 1992, 268, 2416–2419. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Wilkins, O.; Keeler, A.M.; Flotte, T.R. CAR T-Cell Therapy: Progress and Prospects. Hum. Gene Ther. Methods 2017, 28, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Adams, S.; Howe, S.J.; Al Ghonaium, A.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Long-Term Persistence of a Polyclonal T Cell Repertoire After Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci. Transl. Med. 2011, 3, 97ra79. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; Basile, G.D.S.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.-L.; et al. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Aiuti, A.; Vai, S.; Mortellaro, A.; Casorati, G.; Ficara, F.; Andolfi, G.; Ferrari, G.; Tabucchi, A.; Carlucci, F.; Ochs, H.D.; et al. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat. Med. 2002, 8, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Díez, I.A.; Dewey, R.A.; Böhm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-Cell Gene Therapy for the Wiskott–Aldrich Syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Göhring, G.; Steinemann, D.; et al. Gene Therapy for Wiskott-Aldrich Syndrome--Long-Term Efficacy and Genotoxicity. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kühlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Piras, F.; Riba, M.; Petrillo, C.; Lazarevič, D.; Cuccovillo, I.; Bartolaccini, S.; Stupka, E.; Gentner, B.; Cittaro, D.; Naldini, L.; et al. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol. Med. 2017, 9, 1198–1211. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F.D. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Cattoglio, C.; Pellin, D.; Rizzi, E.; Maruggi, G.; Corti, G.; Miselli, F.; Sartori, D.; Guffanti, A.; Di Serio, C.; Ambrosi, A.; et al. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood 2010, 116, 5507–5517. [Google Scholar] [CrossRef] [PubMed]
- Romano, O.; Peano, C.; Tagliazucchi, G.M.; Petiti, L.; Poletti, V.; Cocchiarella, F.; Rizzi, E.; Severgnini, M.; Cavazza, A.; Rossi, C.; et al. Transcriptional, epigenetic and retroviral signatures identify regulatory regions involved in hematopoietic lineage commitment. Sci. Rep. 2016, 6, 24724. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional Transformation of Hematopoietic Cells by Self-inactivating Lentiviral and Gammaretroviral Vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef]
- Chu, J.I.; Henderson, L.A.; Armant, M.; Male, F.; Dansereau, C.H.; MacKinnon, B.; Burke, C.J.; Cavanaugh, M.E.; London, W.B.; Barlan, I.B.; et al. Gene Therapy Using a Self-Inactivating Lentiviral Vector Improves Clinical and Laboratory Manifestations of Wiskott-Aldrich Syndrome. Blood 2015, 126, 260. [Google Scholar] [CrossRef]
- Kohn, D.B.; Hershfield, M.S.; Puck, J.M.; Aiuti, A.; Blincoe, A.; Gaspar, H.B.; Notarangelo, L.D.; Grunebaum, E. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2019, 143, 852–863. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Wu, X.; Moir, S.; Kardava, L.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Susan, M.; et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra57. [Google Scholar] [CrossRef]
- Kohn, D.B.; Booth, C.; Kang, E.M.; Pai, S.-Y.; Shaw, K.L.; Santilli, G.; Armant, M.; Buckland, K.F.; Choi, U.; De Ravin, S.S.; et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med. 2020, 26, 200–206. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.A.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nat. Cell Biol. 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Río, P.; Navarro, S.; Wang, W.; Sánchez-Domínguez, R.; Pujol, R.M.; Segovia, J.C.; Bogliolo, M.; Merino, E.; Wu, N.; Salgado, R.; et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019, 25, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Hofling, A.A.; Devine, S.; Vogler, C.; Sands, M.S. Human CD34+ hematopoietic progenitor cell-directed lentiviral-mediated gene therapy in a xenotransplantation model of lysosomal storage disease. Mol. Ther. 2004, 9, 856–865. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Coffin, J.M. Retroviridae and their replication. In Virology; Raven Press: New York, NY, USA, 1990; pp. 1347–1500. [Google Scholar]
- Sundquist, W.I.; Kräusslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser-Pornillos, B.K.; Yeager, M. Atomic-level modelling of the HIV capsid. Nat. Cell Biol. 2011, 469, 424–427. [Google Scholar] [CrossRef]
- Temin, H.M. Homology between RNA from Rous Sarcoma Virus and DNA from Rous Sarcoma Virus-Infected Cells. Proc. Natl. Acad. Sci. USA 1964, 52, 323–329. [Google Scholar] [CrossRef]
- Hughes, S.H.; Shank, P.R.; Spector, D.H.; Kung, H.-J.; Bishop, J.; Varmus, H.E.; Vogt, P.K.; Breitman, M.L. Proviruses of avian sarcoma virus are terminally redundant, co-extensive with unintegrated linear DNA and integrated at many sites. Cell 1978, 15, 1397–1410. [Google Scholar] [CrossRef]
- Hughes, S.H.; Mutschler, A.; Bishop, J.M.; Varmus, H.E. A Rous sarcoma virus provirus is flanked by short direct repeats of a cellular DNA sequence present in only one copy prior to integration. Proc. Natl. Acad. Sci. USA 1981, 78, 4299–4303. [Google Scholar] [CrossRef] [PubMed]
- Majors, J.E.; Swanstrom, R.; DeLorbe, W.J.; Payne, G.S.; Hughes, S.H.; Ortíz, S.; Quintrell, N.; Bishop, J.M.; Varmus, H.E. DNA Intermediates in the Replication of Retroviruses Are Structurally (and Perhaps Functionally) Related to Transposable Elements. Cold Spring Harb. Symp. Quant. Biol. 1981, 45, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Fassati, A.; Goff, S.P. Characterization of Intracellular Reverse Transcription Complexes of Human Immunodeficiency Virus Type 1. J. Virol. 2001, 75, 3626–3635. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: Studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [CrossRef]
- Burdick, R.C.; Delviks-Frankenberry, K.; Chen, J.; Janaka, S.K.; Sastri, J.; Hu, W.-S.; Pathak, V.K. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017, 13, e1006570. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1–8. [Google Scholar] [CrossRef]
- Jang, S.; Cook, N.J.; Pye, V.E.; Bedwell, G.J.; Dudek, A.M.; Singh, P.K.; Cherepanov, P.; Engelman, A.N. Differential role for phosphorylation in alternative polyadenylation function versus nuclear import of SR-like protein CPSF6. Nucleic Acids Res. 2019, 47, 4663–4683. [Google Scholar] [CrossRef]
- De Iaco, A.; Santoni, F.; Vannier, A.; Guipponi, M.; Antonarakis, S.E.; Luban, J. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 2013, 10, 20. [Google Scholar] [CrossRef]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 Capsid-Cyclophilin Interactions Determine Nuclear Import Pathway, Integration Targeting and Replication Efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible Use of Nuclear Import Pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; et al. Retroviral DNA Integration: Viral and Cellular Determinants of Target-Site Selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A. The benefits of integration. Clin. Microbiol. Infect. 2016, 22, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Paillart, J.-C.; Shehu-Xhilaga, M.; Marquet, R.; Mak, J. Dimerization of retroviral RNA genomes: An inseparable pair. Nat. Rev. Genet. 2004, 2, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.; Kleiman, L. Primer tRNAs for reverse transcription. J. Virol. 1997, 71, 8087–8095. [Google Scholar] [CrossRef]
- Frankel, A.D.; Young, J.A.T. HIV-1: Fifteen Proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Telesnitsky, A.; Goff, S. Reverse Transcriptase and the Generation of Retroviral DNA. In Retroviruses; Cold Spring Harbor Laboratory Press, Cold Spring Harbor: Long Island, NY, USA, 1997; ISBN 0879695714. [Google Scholar]
- Charneau, P.; Alizon, M.; Clavel, F. A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J. Virol. 1992, 66, 2814–2820. [Google Scholar] [CrossRef]
- Fischer, U.; Huber, J.; Boelens, W.C.; Mattajt, L.W.; Lührmann, R. The HIV-1 Rev Activation Domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 1995, 82, 475–483. [Google Scholar] [CrossRef]
- Gilboa, E.; Mitra, S.W.; Goff, S.; Baltimore, D. A detailed model of reverse transcription and tests of crucial aspects. Cell 1979, 18, 93–100. [Google Scholar] [CrossRef]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-Inactivating Lentivirus Vector for Safe and Efficient In Vivo Gene Delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef]
- Wyatt, R. The HIV-1 Envelope Glycoproteins: Fusogens, Antigens, and Immunogens. Science 1998, 280, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G.; Beverley, P.C.L.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; A Weiss, R. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nat. Cell Biol. 1984, 312, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Klatzmann, D.; Champagne, E.; Chamaret, S.; Gruest, J.; Guetard, D.; Hercend, T.; Gluckman, J.-C.; Montagnier, L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nat. Cell Biol. 1984, 312, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Marzio, P.D.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nat. Cell Biol. 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Doranz, B.J.; Rucker, J.; Yi, Y.; Smyth, R.J.; Samson, M.; Peiper, S.C.; Parmentier, M.; Collman, R.G.; Doms, R.W. A Dual-Tropic Primary HIV-1 Isolate That Uses Fusin and the β-Chemokine Receptors CKR-5, CKR-3, and CKR-2b as Fusion Cofactors. Cell 1996, 85, 1149–1158. [Google Scholar] [CrossRef]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1, MIP-1 Receptor as a Fusion Cofactor for Macrophage-Tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.S.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nat. Cell Biol. 1996, 381, 667–673. [Google Scholar] [CrossRef]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.-L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.-M.; Clark-Lewis, I.; Legler, D.F.; et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nat. Cell Biol. 1996, 382, 833–835. [Google Scholar] [CrossRef]
- Abrahamyan, L.G.; Markosyan, R.M.; Moore, J.P.; Cohen, F.S.; Melikyan, G.B. Human Immunodeficiency Virus Type 1 Env with an Intersubunit Disulfide Bond Engages Coreceptors but Requires Bond Reduction after Engagement to Induce Fusion. J. Virol. 2003, 77, 5829–5836. [Google Scholar] [CrossRef]
- Koshiba, T.; Chan, D.C. The Prefusogenic Intermediate of HIV-1 gp41 Contains Exposed C-peptide Regions. J. Biol. Chem. 2002, 278, 7573–7579. [Google Scholar] [CrossRef]
- Si, Z.; Madani, N.; Cox, J.M.; Chruma, J.J.; Klein, J.C.; Schön, A.; Phan, N.; Wang, L.; Biorn, A.C.; Cocklin, S.; et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 5036–5041. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core Structure of gp41 from the HIV Envelope Glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Mol. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nat. Cell Biol. 1997, 387, 426–430. [Google Scholar] [CrossRef]
- Sengupta, P.; Seo, A.Y.; Pasolli, H.A.; Song, Y.E.; Johnson, M.C.; Lippincott-Schwartz, J. A lipid-based partitioning mechanism for selective incorporation of proteins into membranes of HIV particles. Nat. Cell Biol. 2019, 21, 452–461. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Hildreth, J.E.K. Evidence for Budding of Human Immunodeficiency Virus Type 1 Selectively from Glycolipid-Enriched Membrane Lipid Rafts. J. Virol. 2000, 74, 3264–3272. [Google Scholar] [CrossRef]
- Varior-Krishnan, G.; Trescol-Biémont, M.C.; Naniche, D.; Rabourdin-Combe, C.; Gerlier, D. Glycosyl-phosphatidylinositol-anchored and transmembrane forms of CD46 display similar measles virus receptor properties: Virus binding, fusion, and replication; down-regulation by hemagglutinin; and virus uptake and endocytosis for antigen presentation by major histocompatibility complex class II molecules. J. Virol. 1994, 68, 7891–7899. [Google Scholar] [CrossRef]
- Komura, N.; Suzuki, K.G.N.; Ando, H.; Konishi, M.; Koikeda, M.; Imamura, A.; Chadda, R.; Fujiwara, T.K.; Tsuboi, H.; Sheng, R.; et al. Raft-based interactions of gangliosides with a GPI-anchored receptor. Nat. Chem. Biol. 2016, 12, 402–410. [Google Scholar] [CrossRef]
- Tsui-Pierchala, B.A.; Encinas, M.; Milbrandt, J.; Johnson, E.M. Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002, 25, 412–417. [Google Scholar] [CrossRef]
- Miyagawa-Yamaguchi, A.; Kotani, N.; Honke, K. Expressed Glycosylphosphatidylinositol-Anchored Horseradish Peroxidase Identifies Co-Clustering Molecules in Individual Lipid Raft Domains. PLoS ONE 2014, 9, e93054. [Google Scholar] [CrossRef]
- Marschang, P.; Sodroski, J.; Würzner, R.; Dierich, M.P. Decay-accelerating factor (CD55) protects human immunodeficiency virus type 1 from inactivation by human complement. Eur. J. Immunol. 1995, 25, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Bohanakashtan, O. Cell signals transduced by complement. Mol. Immunol. 2004, 41, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Saifuddin, M.; Holguin, M.H.; Parker, C.J.; Hedayati, T.; Atkinson, J.P.; Spear, G.T. Human immunodeficiency virus type 1 incorporates both glycosyl phosphatidylinositol-anchored CD55 and CD59 and integral membrane CD46 at levels that protect from complement-mediated destruction. J. Gen. Virol. 1997, 78, 1907–1911. [Google Scholar] [CrossRef] [PubMed]
- Amet, T.; Lan, J.; Shepherd, N.; Yang, K.; Byrd, D.; Xing, Y.; Yu, Q. Glycosylphosphatidylinositol Anchor Deficiency Attenuates the Production of Infectious HIV-1 and Renders Virions Sensitive to Complement Attack. AIDS Res. Hum. Retrovir. 2016, 32, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Wyma, D.J.; Kotov, A.; Aiken, C. Evidence for a Stable Interaction of gp41 with Pr55Gag in Immature Human Immunodeficiency Virus Type 1 Particles. J. Virol. 2000, 74, 9381–9387. [Google Scholar] [CrossRef]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef]
- Wang, Z.; Schey, K.L. Proteomic Analysis of Lipid Raft-Like Detergent-Resistant Membranes of Lens Fiber Cells. Investig. Opthalmol. Vis. Sci. 2015, 56, 8349–8360. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Schlesinger, M.J. Fatty acid binding to vesicular stomatitis virus glycoprotein: A new type of post-translational modification of the viral glycoprotein. Cell 1979, 17, 813–819. [Google Scholar] [CrossRef]
- Schmidt, M.F. Acylation of virol. spike glycoproteins: A feature of enveloped RNA viruses. Virology 1982, 116, 327–338. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Bracha, M.; Schlesinger, M.J. Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins. Proc. Natl. Acad. Sci. USA 1979, 76, 1687–1691. [Google Scholar] [CrossRef]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [PubMed]
- Lentz, T.L.; Burrage, T.G.; Smith, A.L.; Crick, J.; Tignor, G.H. Is the acetylcholine receptor a rabies virus receptor? Science 1982, 215, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Thoulouze, M.-I.; Lafage, M.; Schachner, M.; Hartmann, U.; Cremer, H.; Lafon, M. The Neural Cell Adhesion Molecule Is a Receptor for Rabies Virus. J. Virol. 1998, 72, 7181–7190. [Google Scholar] [CrossRef]
- Tuffereau, C.; Bénéjean, J.; Blondel, D.; Kieffer, B.; Flamand, A. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. EMBO J. 1998, 17, 7250–7259. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Liu, R.; Shuai, L.; Wang, X.; Luo, J.; Wang, C.; Chen, W.; Wang, X.; Ge, J.; et al. Metabotropic glutamate receptor subtype 2 is a cellular receptor for rabies virus. PLoS Pathog. 2018, 14, e1007189. [Google Scholar] [CrossRef]
- Bartz, R.; Firsching, R.; Ter Meulen, V.; Schneider-Schaulies, J.; Rima, B. Differential receptor usage by measles virus strains. J. Gen. Virol. 1998, 79, 1015–1025. [Google Scholar] [CrossRef]
- Hsu, E.C.; Sarangi, F.; Iorio, C.; Sidhu, M.S.; Udem, S.A.; Dillehay, D.L.; Xu, W.; Rota, P.A.; Bellini, W.J.; Richardson, C.D. A Single Amino Acid Change in the Hemagglutinin Protein of Measles Virus Determines Its Ability to Bind CD46 and Reveals Another Receptor on Marmoset B Cells. J. Virol. 1998, 72, 2905–2916. [Google Scholar] [CrossRef]
- Buckland, R.; Wild, T.F. Is CD46 the cellular receptor for measles virus? Virus Res. 1997, 48, 1–9. [Google Scholar] [CrossRef]
- Hsu, E.C.; Iorioab, C.; Sarangiab, F.; AyeKhineab, A.; Richardson, C.D. CDw150(SLAM) Is a Receptor for a Lymphotropic Strain of Measles Virus and May Account for the Immunosuppressive Properties of This Virus. Virology 2001, 279, 9–21. [Google Scholar] [CrossRef]
- Murabayashi, N.; Kurita-Taniguchi, M.; Ayata, M.; Matsumoto, M.; Ogura, H.; Seya, T. Susceptibility of human dendritic cells (DCs) to measles virus (MV) depends on their activation stages in conjunction with the level of CDw150: Role of Toll stimulators in DC maturation and MV amplification. Microbes Infect. 2002, 4, 785–794. [Google Scholar] [CrossRef]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. SLAM (CDw150) is a cellular receptor for measles virus. Nat. Cell Biol. 2000, 406, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Verhoeyen, E.; Cosset, F.-L. Surface-engineering of lentiviral vectors. J. Gene Med. 2004, 6, S83–S94. [Google Scholar] [CrossRef] [PubMed]
- Lévy, C.; Amirache, F.; Girard-Gagnepain, A.; Frecha, C.; Roman-Rodríguez, F.J.; Bernadin, O.; Costa, C.; Nègre, D.; Gutierrez-Guerrero, A.; Vranckx, L.S.; et al. Measles virus envelope pseudotyped lentiviral vectors transduce quiescent human HSCs at an efficiency without precedent. Blood Adv. 2017, 1, 2088–2104. [Google Scholar] [CrossRef] [PubMed]
- Witting, S.R.; Vallanda, P.; Gamble, A.L. Characterization of a third generation lentiviral vector pseudotyped with Nipah virus envelope proteins for endothelial cell transduction. Gene Ther. 2013, 20, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Palomares, K.; Vigant, F.; Van Handel, B.; Pernet, O.; Chikere, K.; Hong, P.; Sherman, S.P.; Patterson, M.; An, D.S.; Lowry, W.E.; et al. Nipah Virus Envelope-Pseudotyped Lentiviruses Efficiently Target ephrinB2-Positive Stem Cell Populations In Vitro and Bypass the Liver Sink When Administered In Vivo. J. Virol. 2012, 87, 2094–2108. [Google Scholar] [CrossRef]
- Wintachai, P.; Wikan, N.; Kuadkitkan, A.; Jaimipuk, T.; Ubol, S.; Pulmanausahakul, R.; Auewarakul, P.; Kasinrerk, W.; Weng, W.-Y.; Panyasrivanit, M.; et al. Identification of prohibitin as a Chikungunya virus receptor protein. J. Med. Virol. 2012, 84, 1757–1770. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nat. Cell Biol. 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Matusali, G.; Colavita, F.; Bordi, L.; Lalle, E.; Ippolito, G.; Capobianchi, M.R.; Castilletti, C. Tropism of the Chikungunya Virus. Viruses 2019, 11, 175. [Google Scholar] [CrossRef]
- Salvador, B.; Zhou, Y.; Michault, A.; Muench, M.O.; Simmons, G. Characterization of Chikungunya pseudotyped viruses: Identification of refractory cell lines and demonstration of cellular tropism differences mediated by mutations in E1 glycoprotein. Virology 2009, 393, 33–41. [Google Scholar] [CrossRef]
- Wang, K.S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 1992, 66, 4992–5001. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.P.; Hanna, S.L.; Spiridigliozzi, A.; Wannissorn, N.; Beiting, D.P.; Ross, S.R.; Hardy, R.W.; Bambina, S.A.; Heise, M.T.; Cherry, S. Natural Resistance-Associated Macrophage Protein Is a Cellular Receptor for Sindbis Virus in Both Insect and Mammalian Hosts. Cell Host Microbe 2011, 10, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Elegheert, J.; Behiels, E.; Bishop, B.; Scott, S.; Woolley, R.E.; Griffiths, S.C.; Byrne, E.F.; Chang, V.T.; Stuart, D.I.; Jones, E.Y.; et al. Lentiviral transduction of mammalian cells for fast, scalable and high-level production of soluble and membrane proteins. Nat. Protoc. 2018, 13, 2991–3017. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, K.; Mirnikjoo, B.; Schroit, A.J. Regulated Externalization of Phosphatidylserine at the Cell Surface. J. Biol. Chem. 2007, 282, 18357–18364. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Coil, D.A.; Miller, A.D. Phosphatidylserine Is Not the Cell Surface Receptor for Vesicular Stomatitis Virus. J. Virol. 2004, 78, 10920–10926. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef]
- Go, G.-W.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J. Biol. Med. 2012, 85, 19–28. [Google Scholar]
- Sun, X.; Roth, S.L.; Bialecki, M.A.; Whittaker, G.R. Internalization and fusion mechanism of vesicular stomatitis virus and related rhabdoviruses. Futur. Virol. 2010, 5, 85–96. [Google Scholar] [CrossRef]
- Clague, M.J.; Schoch, C.; Zech, L.; Blumenthal, R. Gating kinetics of pH-activated membrane fusion of vesicular stomatitis virus with cells: Stopped-flow measurements by dequenching of octadecylrhodamine fluorescence. Biochemistry 1990, 29, 1303–1308. [Google Scholar] [CrossRef]
- Paternostre, M.-T.; Lowy, R.J.; Blumenthal, R. pH-dependent fusion of reconstituted vesicular stomatitis virus envelopes with vero cells Measurement by dequenching of fluorescence. FEBS Lett. 1989, 243, 251–258. [Google Scholar] [CrossRef]
- Sun, X.; Yau, V.K.; Briggs, B.J.; Whittaker, G.R. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 2005, 338, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Albertini, A.A.; Baquero, E.; Ferlin, A.; Gaudin, Y. Molecular and Cellular Aspects of Rhabdovirus Entry. Viruses 2012, 4, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Jenni, S.; Stanifer, M.L.; Roth, E.; Whelan, S.P.J.; Van Oijen, A.M.; Harrison, S.C. Mechanism of membrane fusion induced by vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. USA 2017, 114, E28–E36. [Google Scholar] [CrossRef] [PubMed]
- Cantore, A.; Nair, N.; Della Valle, P.; Di Matteo, M.; Màtrai, J.; Sanvito, F.; Brombin, C.; Di Serio, C.; D’Angelo, A.; Chuah, M.K.L.; et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood 2012, 120, 4517–4520. [Google Scholar] [CrossRef]
- Cantore, A.A.; Ranzani, M.M.; Bartholomae, C.C.; Volpin, M.M.; Valle, P.D.P.; Sanvito, F.F.; Sergi, L.S.L.; Gallina, P.P.; Benedicenti, F.F.; Bellinger, D.D.; et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 2015, 7, 277ra28. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kimura, T.; Haga, K.; Kasahara, N.; Anton, P.A.; McGowan, I. Effective In Vivo and Ex Vivo gene transfer to intestinal mucosa by VSV-G-pseudotyped lentiviral vectors. BMC Gastroenterol. 2010, 10, 44. [Google Scholar] [CrossRef]
- Kawabata, K.; Migita, M.; Mochizuki, H.; Miyake, K.; Igarashi, T.; Fukunaga, Y.; Shimada, T. Ex vivo cell-mediated gene therapy for metachromatic leukodystrophy using neurospheres. Brain Res. 2006, 1094, 13–23. [Google Scholar] [CrossRef]
- Suzuki, A.; Obi, K.; Urabe, T.; Hayakawa, H.; Yamada, M.; Kaneko, S.; Onodera, M.; Mizuno, Y.; Mochizuki, H. Feasibility of Ex Vivo gene therapy for neurological disorders using the new retroviral vector GCDNsap packaged in the vesicular stomatitis virus G protein. J. Neurochem. 2002, 82, 953–960. [Google Scholar] [CrossRef][Green Version]
- Mazarakis, N.D.; Azzouz, M.; Rohll, J.B.; Ellard, F.M.; Wilkes, F.J.; Olsen, A.L.; Carter, E.E.; Barber, R.D.; Baban, D.F.; Kingsman, S.M.; et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 2001, 10, 2109–2121. [Google Scholar] [CrossRef]
- Piccinotti, S.; Whelan, S.P. Rabies Internalizes into Primary Peripheral Neurons via Clathrin Coated Pits and Requires Fusion at the Cell Body. PLoS Pathog. 2016, 12, e1005753. [Google Scholar] [CrossRef] [PubMed]
- Burrage, T.G.; Tignor, G.H.; Smith, A.L. Rabies virus binding at neuromuscular junctions. Virus Res. 1985, 2, 273–289. [Google Scholar] [CrossRef]
- Tuffereau, C.; Schmidt, K.; Langevin, C.; Lafay, F.; DeChant, G.; Koltzenburg, M. The Rabies Virus Glycoprotein Receptor p75NTR Is Not Essential for Rabies Virus Infection. J. Virol. 2007, 81, 13622–13630. [Google Scholar] [CrossRef] [PubMed]
- Kyrkanides, S.; Miller, J.H.; Brouxhon, S.M.; Olschowka, J.A.; Federoff, H.J. β-hexosaminidase lentiviral vectors: Transfer into the CNS via systemic administration. Mol. Brain Res. 2005, 133, 286–298. [Google Scholar] [CrossRef]
- Chang, A.; Dutch, R.E. Paramyxovirus Fusion and Entry: Multiple Paths to a Common End. Viruses 2012, 4, 613–636. [Google Scholar] [CrossRef]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 112. [Google Scholar] [CrossRef]
- Yanagi, Y.; Takeda, M.; Ohno, S. Measles virus: Cellular receptors, tropism and pathogenesis. J. Gen. Virol. 2006, 87, 2767–2779. [Google Scholar] [CrossRef]
- Dörig, R.E.; Marcil, A.; Chopra, A.; Richardson, C.D. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 1993, 75, 295–305. [Google Scholar] [CrossRef]
- Naniche, D.; Varior-Krishnan, G.; Cervoni, F.; Wild, T.F.; Rossi, B.; Rabourdin-Combe, C.; Gerlier, D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 1993, 67, 6025–6032. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Post, T.W.; Atkinson, J.P. Membrane cofactor protein (MCP or CD46): Newest member of the regulators of complement activation gene cluster. Annu. Rev. Immunol. 1991, 9, 431–455. [Google Scholar] [CrossRef]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.-T.; Sisson, G.; Tsao, M.-S.; Richardson, C.D. Tumor Cell Marker PVRL4 (Nectin 4) Is an Epithelial Cell Receptor for Measles Virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef]
- Mühlebach, M.D.; Mateo, M.; Sinn, P.L.; Prüfer, S.; Uhlig, K.M.; Leonard, V.H.J.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nat. Cell Biol. 2011, 480, 530–533. [Google Scholar] [CrossRef] [PubMed]
- DeRycke, M.S.; Pambuccian, S.E.; Gilks, C.B.; Kalloger, S.E.; Ghidouche, A.; Lopez, M.; Bliss, R.L.; Geller, M.A.; Argenta, P.A.; Harrington, K.M.; et al. Nectin 4 Overexpression in Ovarian Cancer Tissues and Serum. Am. J. Clin. Pathol. 2010, 134, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Takano, A.; Ishikawa, N.; Nishino, R.; Masuda, K.; Yasui, W.; Inai, K.; Nishimura, H.; Ito, H.; Nakayama, H.; Miyagi, Y.; et al. Identification of Nectin-4 Oncoprotein as a Diagnostic and Therapeutic Target for Lung Cancer. Cancer Res. 2009, 69, 6694–6703. [Google Scholar] [CrossRef] [PubMed]
- Vongpunsawad, S.; Oezgun, N.; Braun, W.; Cattaneo, R. Selectively Receptor-Blind Measles Viruses: Identification of Residues Necessary for SLAM or CD46-Induced Fusion and Their Localization on a New Hemagglutinin Structural Model. J. Virol. 2004, 78, 302–313. [Google Scholar] [CrossRef]
- Nakamura, T.; Peng, K.-W.; Harvey, M.; Greiner, S.; Lorimer, I.A.J.; James, C.D.; Russell, S.J. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 2005, 23, 209–214. [Google Scholar] [CrossRef]
- Münch, R.C.; Mühlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Plückthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An Efficient Targeting Domain for Lentiviral Vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef]
- De Swart, R.L.; Yüksel, S.; Osterhaus, A.D.M.E. Relative Contributions of Measles Virus Hemagglutinin- and Fusion Protein-Specific Serum Antibodies to Virus Neutralization. J. Virol. 2005, 79, 11547–11551. [Google Scholar] [CrossRef]
- Kneissl, S.; Abel, T.; Rasbach, A.; Brynza, J.; Schneider-Schaulies, J.; Buchholz, C.J. Measles Virus Glycoprotein-Based Lentiviral Targeting Vectors That Avoid Neutralizing Antibodies. PLoS ONE 2012, 7, e46667. [Google Scholar] [CrossRef]
- Khetawat, D.; Broder, C.C. A Functional Henipavirus Envelope Glycoprotein Pseudotyped Lentivirus Assay System. Virol. J. 2010, 7, 312. [Google Scholar] [CrossRef]
- Bruhn, J.F.; Barnett, K.C.; Bibby, J.; Thomas, J.M.H.; Keegan, R.M.; Rigden, D.J.; Bornholdt, Z.A.; Saphire, E.O. Crystal Structure of the Nipah Virus Phosphoprotein Tetramerization Domain. J. Virol. 2013, 88, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Bender, R.R.; Muth, A.; Schneider, I.C.; Friedel, T.; Hartmann, J.; Plückthun, A.; Maisner, A.; Buchholz, C.J. Receptor-Targeted Nipah Virus Glycoproteins Improve Cell-Type Selective Gene Delivery and Reveal a Preference for Membrane-Proximal Cell Attachment. PLoS Pathog. 2016, 12, e1005641. [Google Scholar] [CrossRef] [PubMed]
- Enkirch, T.; Kneissl, S.; Hoyler, B.; Ungerechts, G.; Stremmel, W.; Buchholz, C.J.; Springfeld, C. Targeted lentiviral vectors pseudotyped with the Tupaia paramyxovirus glycoproteins. Gene Ther. 2012, 20, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.; Maisner, A.; Mühlebach, M.D.; Koehl, U.; Grez, M.; Cattaneo, R.; Cichutek, K.; Buchholz, C.J. Targeted Cell Entry of Lentiviral Vectors. Mol. Ther. 2008, 16, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Bonaparte, M.I.; Dimitrov, D.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Wang, L.-F.; Eaton, B.T.; Broder, C.C. From the Cover: Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [PubMed]
- Negrete, O.A.; Wolf, M.C.; Aguilar, H.C.; Enterlein, S.; Wang, W.; Mühlberger, E.; Su, S.V.; Bertolotti-Ciarlet, A.; Flick, R.; Lee, B. Two Key Residues in EphrinB3 Are Critical for Its Use as an Alternative Receptor for Nipah Virus. PLoS Pathog. 2006, 2, 0078–0086. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Garcia-Cardena, G.; Hayashid, S.-I.; Geretya, S.; Asaharad, T.; Stavrakisb, G.; Isnerd, J.; Folkman, J.; Gimbrone, M.A.; Anderson, D.J. Expression of EphrinB2 Identifies a Stable Genetic Difference Between Arterial and Venous Vascular Smooth Muscle as Well as Endothelial Cells, and Marks Subsets of Microvessels at Sites of Adult Neovascularization. Dev. Biol. 2001, 230, 139–150. [Google Scholar] [CrossRef]
- Foo, S.S.; Turner, C.J.; Adams, S.; Compagni, A.; Aubyn, D.; Kogata, N.; Lindblom, P.; Shani, M.; Zicha, D.; Adams, R.H. Ephrin-B2 Controls Cell Motility and Adhesion during Blood-Vessel-Wall Assembly. Cell 2006, 124, 161–173. [Google Scholar] [CrossRef]
- Martowicz, A.; Seeber, A.; Untergasser, G. The role of EpCAM in physiology and pathology of the epithelium. Histol. Histopathol. 2015, 31, 349–355. [Google Scholar]
- Wengler, G.; Koschinski, A.; Wengler, G.; Dreyer, F. Entry of alphaviruses at the plasma membrane converts the viral surface proteins into an ion-permeable pore that can be detected by electrophysiological analyses of whole-cell membrane currents. J. Gen. Virol. 2003, 84, 173–181. [Google Scholar] [CrossRef]
- Lee, R.C.H.; Hapuarachchi, H.C.; Chen, K.C.; Hussain, K.M.; Chen, H.; Low, S.L.; Ng, L.C.; Lin, R.; Ng, M.M.-L.; Chu, J.J.H. Mosquito Cellular Factors and Functions in Mediating the Infectious entry of Chikungunya Virus. PLoS Negl. Trop. Dis. 2013, 7, e2050. [Google Scholar] [CrossRef] [PubMed]
- Van Duijl-Richter, M.K.S.; Hoornweg, T.E.; Rodenhuis-Zybert, I.A.; Smit, J.M. Early Events in Chikungunya Virus Infection—From Virus CellBinding to Membrane Fusion. Viruses 2015, 7, 3647–3674. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Cheng, R.H.; Olson, N.H.; Peterson, P.; Chase, E.; Kuhn, R.J.; Baker, T.S. Putative receptor binding sites on alphaviruses as visualized by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 1995, 92, 10648–10652. [Google Scholar] [CrossRef] [PubMed]
- Uchime, O.; Fields, W.; Kielian, M. The Role of E3 in pH Protection during Alphavirus Assembly and Exit. J. Virol. 2013, 87, 10255–10262. [Google Scholar] [CrossRef] [PubMed]
- Carleton, M.; Lee, H.; Mulvey, M.; Brown, D.T. Role of glycoprotein PE2 in formation and maturation of the Sindbis virus spike. J. Virol. 1997, 71, 1558–1566. [Google Scholar] [CrossRef]
- Snyder, A.J.; Mukhopadhyay, S. The Alphavirus E3 Glycoprotein Functions in a Clade-Specific Manner. J. Virol. 2012, 86, 13609–13620. [Google Scholar] [CrossRef]
- Sahoo, B.; Chowdary, T.K. Conformational changes in Chikungunya virus E2 protein upon heparan sulfate receptor binding explain mechanism of E2–E1 dissociation during viral entry. Biosci. Rep. 2019, 39, 39. [Google Scholar] [CrossRef]
- Hua, R.L.C.; Hussain, K.M.; Chu, J.J.H.; Lee, C.H.R. Macropinocytosis dependent entry of Chikungunya virus into human muscle cells. PLoS Negl. Trop. Dis. 2019, 13, e0007610. [Google Scholar] [CrossRef]
- Cerny, T.; Schwarz, M.; Lemant, J.; Gérardin, P.; Keller, E. The Range of Neurological Complications in Chikungunya Fever. Neurocrit. Care 2017, 27, 447–457. [Google Scholar] [CrossRef]
- Thiberville, S.-D.; Moyen, N.; Dupuis-Maguiraga, L.; Nougairede, A.; Gould, E.A.; Roques, P.; De Lamballerie, X. Chikungunya fever: Epidemiology, clinical syndrome, pathogenesis and therapy. Antivir. Res. 2013, 99, 345–370. [Google Scholar] [CrossRef]
- Martinez-Pulgarin, D.F.; Chowdhury, F.R.; Villamil-Gómez, W.E.; Rodríguez-Morales, A.J.; Blohm, G.M.; Paniz-Mondolfi, A.E. Ophthalmologic aspects of chikungunya infection. Travel Med. Infect. Dis. 2016, 14, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadou, I.; Dieringer, M.; Poh, X.Y.; Sanchez-Garrido, J.; Gao, Y.; Sgourou, A.; Simmons, L.E.; Mazarakis, N.D. Selective transduction of astrocytic and neuronal CNS subpopulations by lentiviral vectors pseudotyped with Chikungunya virus envelope. Biomaterials 2017, 123, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Piazzi, M.; Raffini, M.; Faenza, I.; Blalock, W. Prohibitin 2: At a communications crossroads. IUBMB Life 2015, 67, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Bentayeb, H.; Aitamer, M.; Petit, B.; Dubanet, L.; Elderwish, S.; Désaubry, L.; De Gramont, A.; Raymond, E.; Olivrie, A.; Abraham, J.; et al. Prohibitin (PHB) expression is associated with aggressiveness in DLBCL and flavagline-mediated inhibition of cytoplasmic PHB functions induces anti-tumor effects. J. Exp. Clin. Cancer Res. 2019, 38, 450. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.M.; Brault, A.C.; Shirako, Y.; Strauss, E.G.; Kang, W.; Strauss, J.H.; Weaver, S.C. Evolutionary Relationships and Systematics of the Alphaviruses. J. Virol. 2001, 75, 10118–10131. [Google Scholar] [CrossRef] [PubMed]
- Khalfaoui, T.; Groulx, J.-F.; Sabra, G.; Guezguez, A.; Basora, N.; Vermette, P.; Beaulieu, J.-F. Laminin Receptor 37/67LR Regulates Adhesion and Proliferation of Normal Human Intestinal Epithelial Cells. PLoS ONE 2013, 8, e74337. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Pesapane, A.; Giudice, V.; Serio, B.; Rossi, F.W.; De Paulis, A.; Selleri, C. 67 kDa laminin receptor (67LR) in normal and neoplastic hematopoietic cells: Is its targeting a feasible approach? Transl. Med. 2016, 15, 8–14. [Google Scholar]
- Berno, V. The 67 kDa laminin receptor increases tumor aggressiveness by remodeling laminin-1. Endocr. Relat. Cancer 2005, 12, 393–406. [Google Scholar] [CrossRef]
- Gruenheid, S.; Canonne-Hergaux, F.; Gauthier, S.; Hackam, D.J.; Grinstein, S.; Gros, P. The Iron Transport Protein NRAMP2 Is an Integral Membrane Glycoprotein That Colocalizes with Transferrin in Recycling Endosomes. J. Exp. Med. 1999, 189, 831–841. [Google Scholar] [CrossRef]
- Gruenheid, S.; Cellier, M.; Vidal, S.; Gros, P. Identification and characterization of a second mouse Nramp gene. Genomics 1995, 25, 514–525. [Google Scholar] [CrossRef]
- Gunshin, H.; MacKenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nat. Cell Biol. 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.M.; Ferreira, D.; Horton, M.; Saad, A.; Tsuruta, H.; Johnston, R.; Klimstra, W.; Ryman, K.; Hernandez, R.; Chiu, W.; et al. Conformational changes in Sindbis virions resulting from exposure to low pH and interactions with cells suggest that cell penetration may occur at the cell surface in the absence of membrane fusion. Virolology 2004, 324, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Vancini, R.; Wang, G.; Ferreira, D.; Hernandez, R.; Brown, D.T. Alphavirus Genome Delivery Occurs Directly at the Plasma Membrane in a Time- and Temperature-Dependent Process. J. Virol. 2013, 87, 4352–4359. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Sawai, K.; Lijima, Y.; Levin, B.; Meruelo, D. Cell-specific targeting of Sindbis virus vectors displaying IgG-binding domains of protein A. Nat. Biotechnol. 1997, 15, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Bristol, G.; Xie, Y.-M.; Kung, S.K.-P.; Chen, I.S.Y. Antibody-Directed Targeting of Retroviral Vectors via Cell Surface Antigens. J. Virol. 2001, 75, 8016–8020. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Xie, Y.; Ringpis, G.-E.; Johnson, M.; Nassanian, H.; Lee, B.; Wu, L.; Chen, I.S.Y. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 2005, 11, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Ku, A.; Xie, Y.; Harui, A.; Kung, S.K.P.; Roth, M.D.; Lee, B.; Chen, I.S.Y. Redirecting Lentiviral Vectors Pseudotyped with Sindbis Virus-Derived Envelope Proteins to DC-SIGN by Modification of N-Linked Glycans of Envelope Proteins. J. Virol. 2010, 84, 6923–6934. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original Virus | Pseudotype | Main Characteristics of the Pseudotyped LV | Natural Cell Tropism | Receptor | Transduction Efficiency | References |
|---|---|---|---|---|---|---|
| Vesicular Stomatitis virus | VSV-G | Quasi-universal tropism, high efficiency | Broad | LDL-R | High | [95,96] |
| Rabies virus | RabV-G | Natural ability to efficiently targets neurons | Neurons | nAChR, CD56, p75NTR, mGluR2 | Up to 50% | [97,98,99,100] |
| Measle virus | H/F | High efficiency, tolerant to peptide insertion, can be neutralized by vaccines | B cells, T cells, Epithelial cells, Dendritic cells, HSPC | CD46, SLAM, nectin-4 | Up to 50–70% | [101,102,103,104,105,106,107,108] |
| Nipah virus | G/F | Low prevalence: low neutralization hazard | Pericytes, tumor endothelium | EphrinB2, EphrinB4 | 20–40% | [109,110] |
| Chickungunya virus | E1/E2 | Versatile basis for engineering/reprograming | Broad | PHB1, Mxra8, integrins, Heparan sulfates | Low on non-adherent cells, high on adherent cells (related to VSV-G) | [111,112,113,114] |
| Sindbis virus | E1/E2 | Versatile basis for engineering/reprograming, Low immunogenicity | Broad | 67LR, NRAMP2 | Variable | [115,116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duvergé, A.; Negroni, M. Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus. Viruses 2020, 12, 1311. https://doi.org/10.3390/v12111311
Duvergé A, Negroni M. Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus. Viruses. 2020; 12(11):1311. https://doi.org/10.3390/v12111311
Chicago/Turabian StyleDuvergé, Alexis, and Matteo Negroni. 2020. "Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus" Viruses 12, no. 11: 1311. https://doi.org/10.3390/v12111311
APA StyleDuvergé, A., & Negroni, M. (2020). Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus. Viruses, 12(11), 1311. https://doi.org/10.3390/v12111311