RNAseq of Deformed Wing Virus and Other Honey Bee-Associated Viruses in Eight Insect Taxa with or without Varroa Infestation




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Setup
2.2. Sample Preparation and RNA Extraction
2.3. cDNA Library Preparation
2.4. Data Quality Control
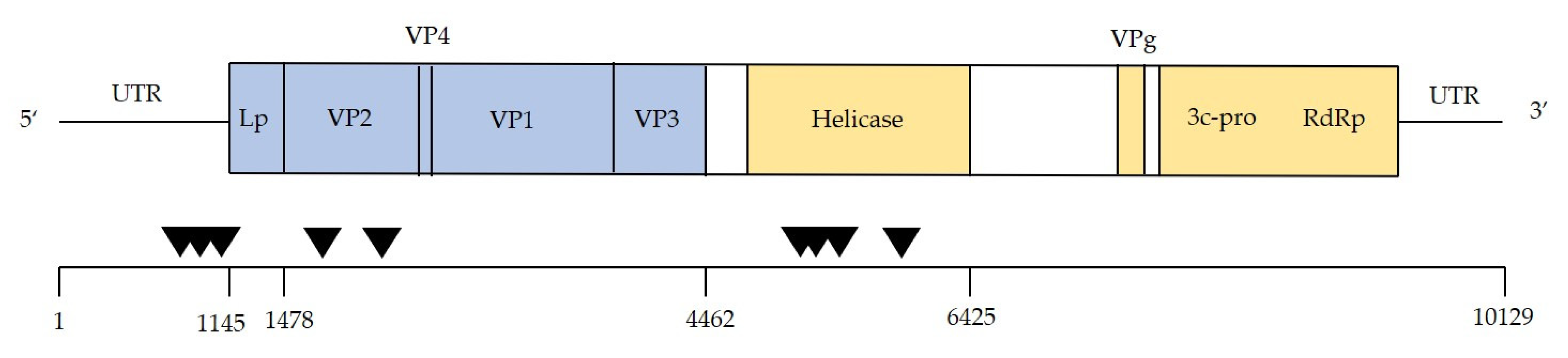
2.5. Identification and Quantification of DWV-Like Reads and Identification of Recombinants
2.6. Determination of the Extent to Which Common DWV Variants Are Shared
2.7. Determination of DWV Quasispecies Diversity
2.8. Identification of Other Honey Bee-Associated Viruses
3. Results
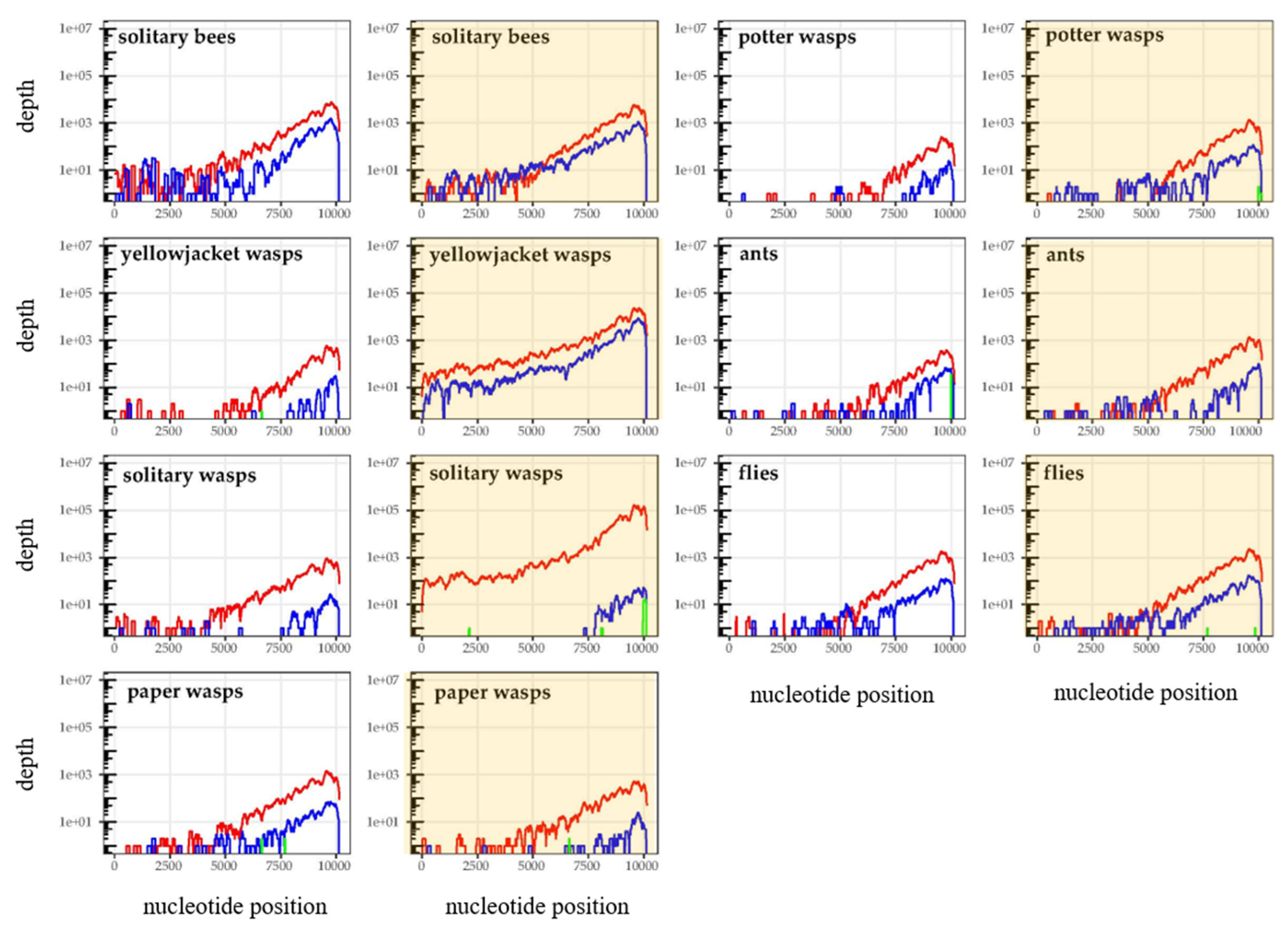
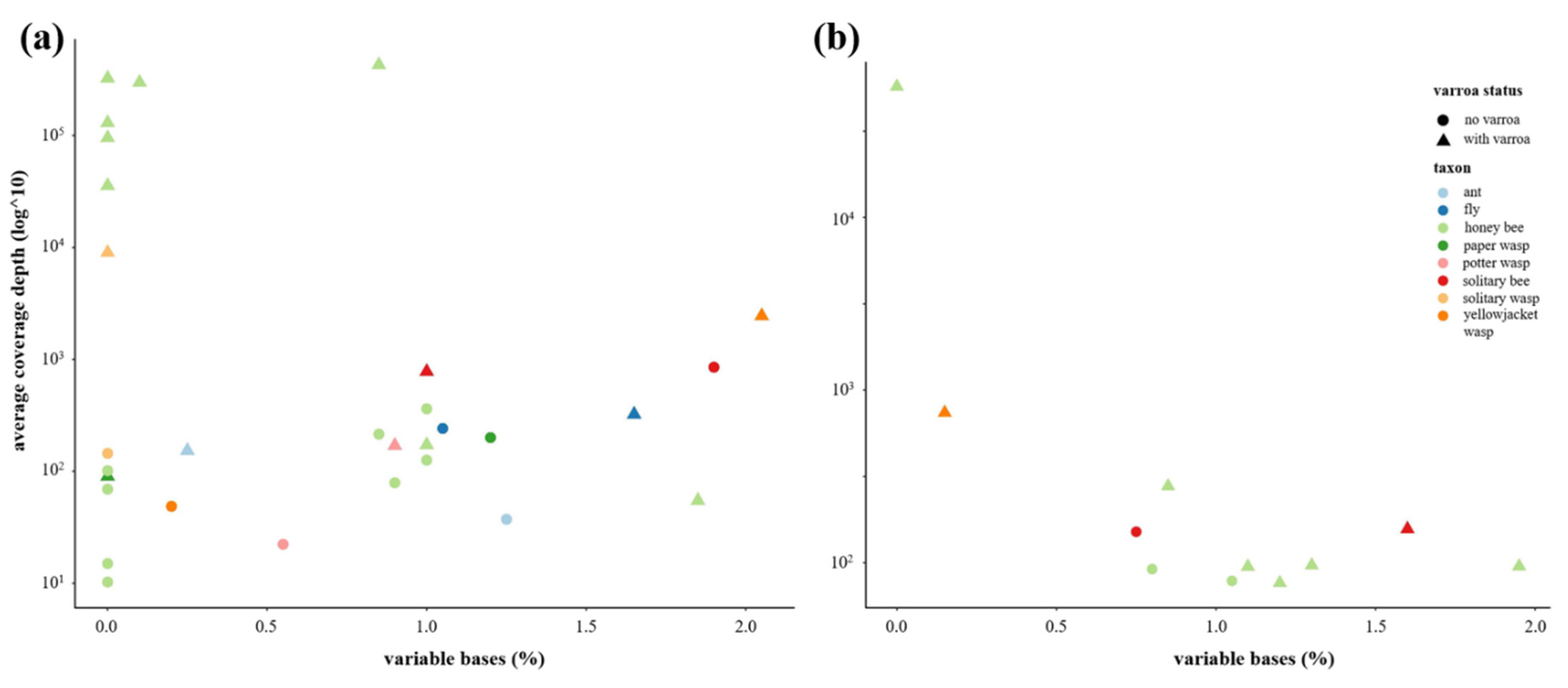
3.1. Sequence Data Statistics and Quality Control
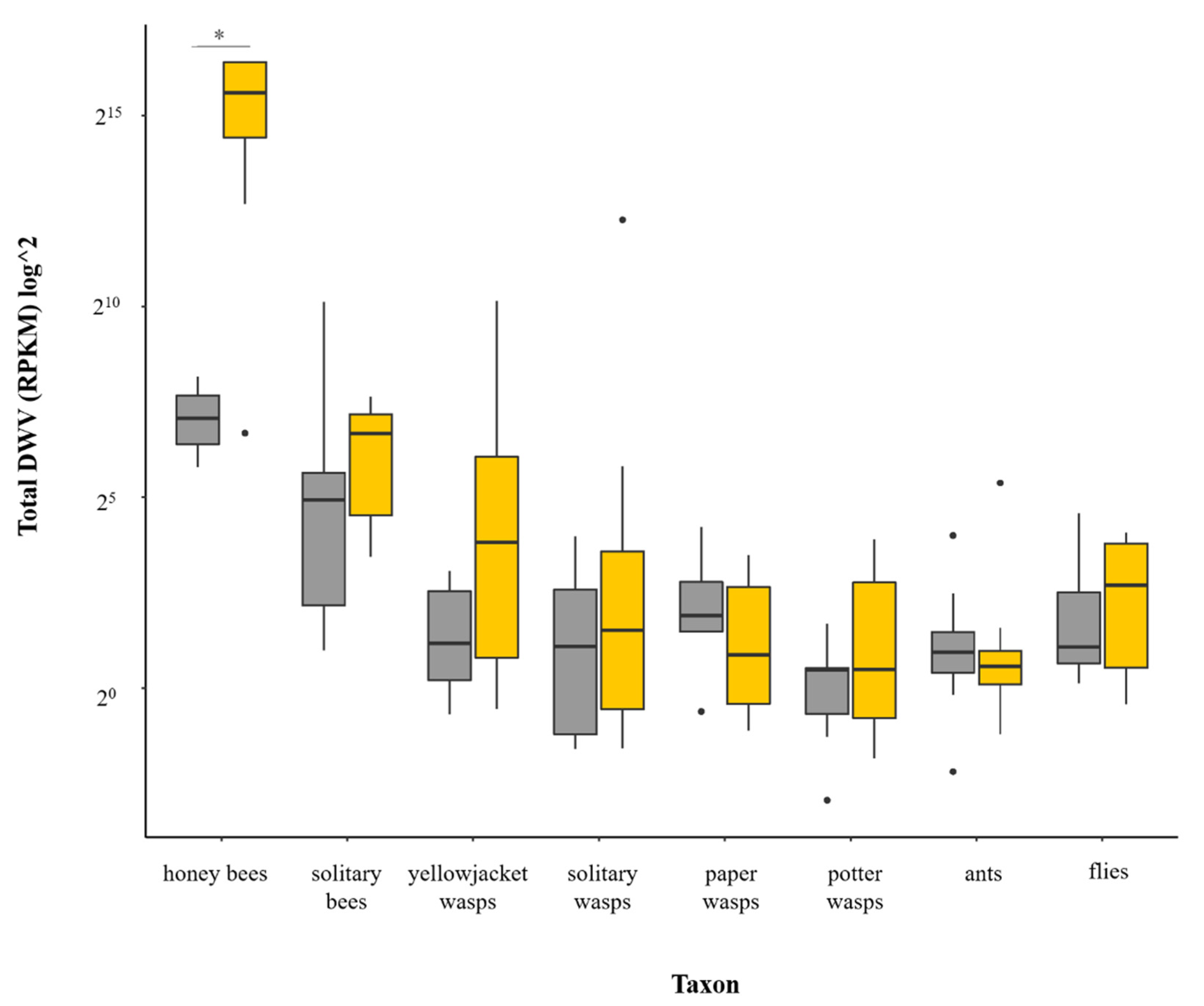
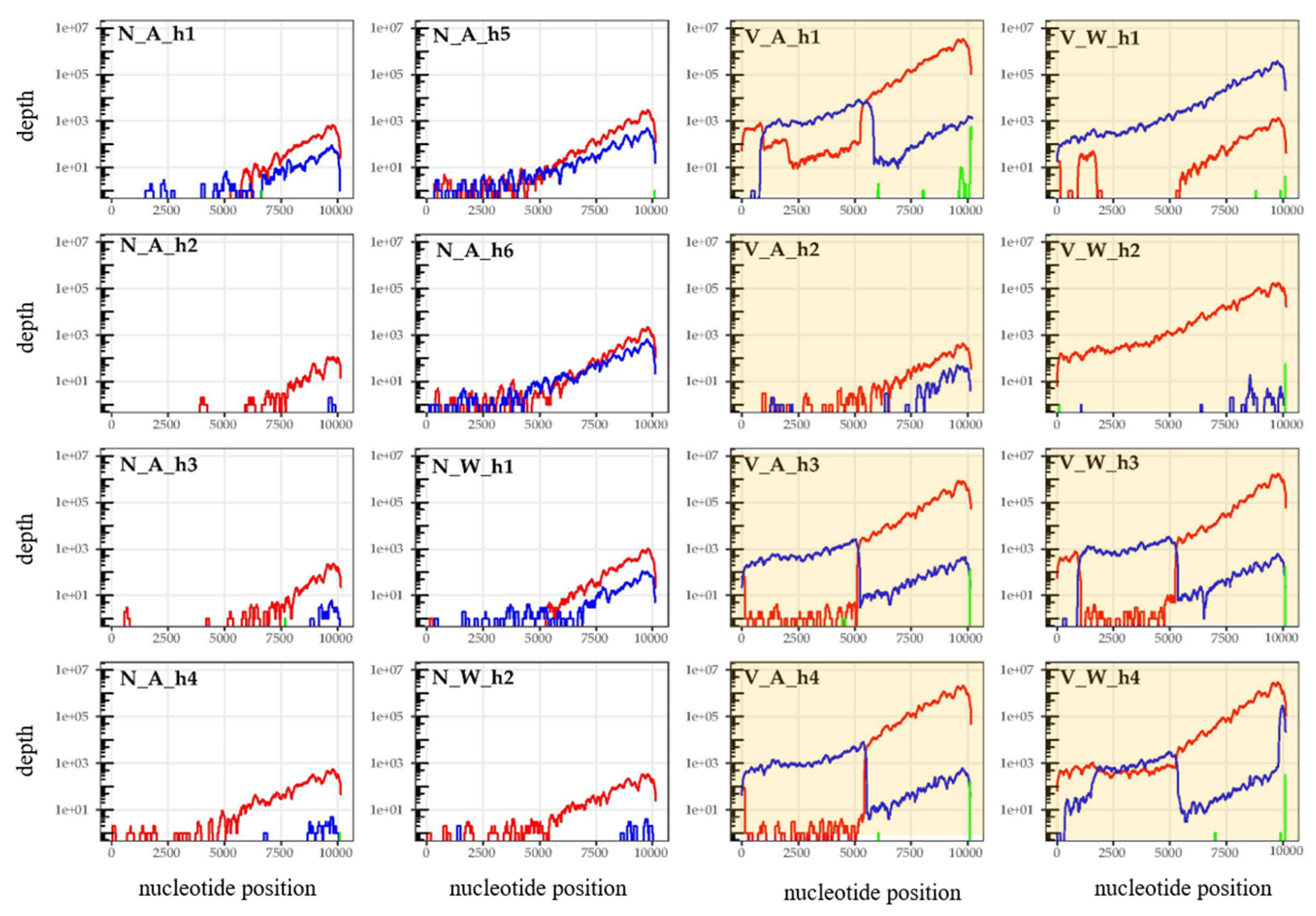
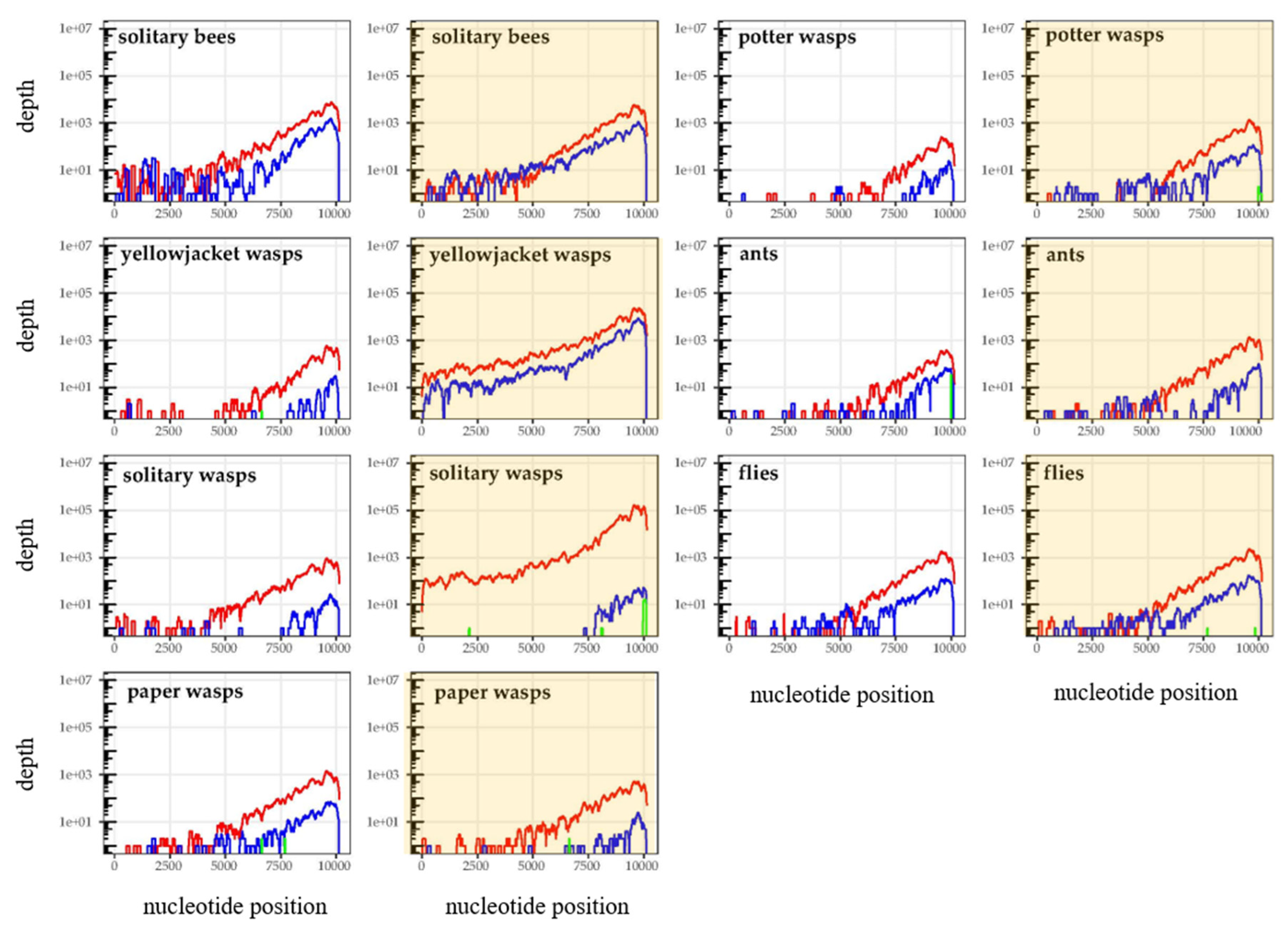
3.2. Is Varroa-Presence Associated with an Increase in DWV in Diverse Taxa?
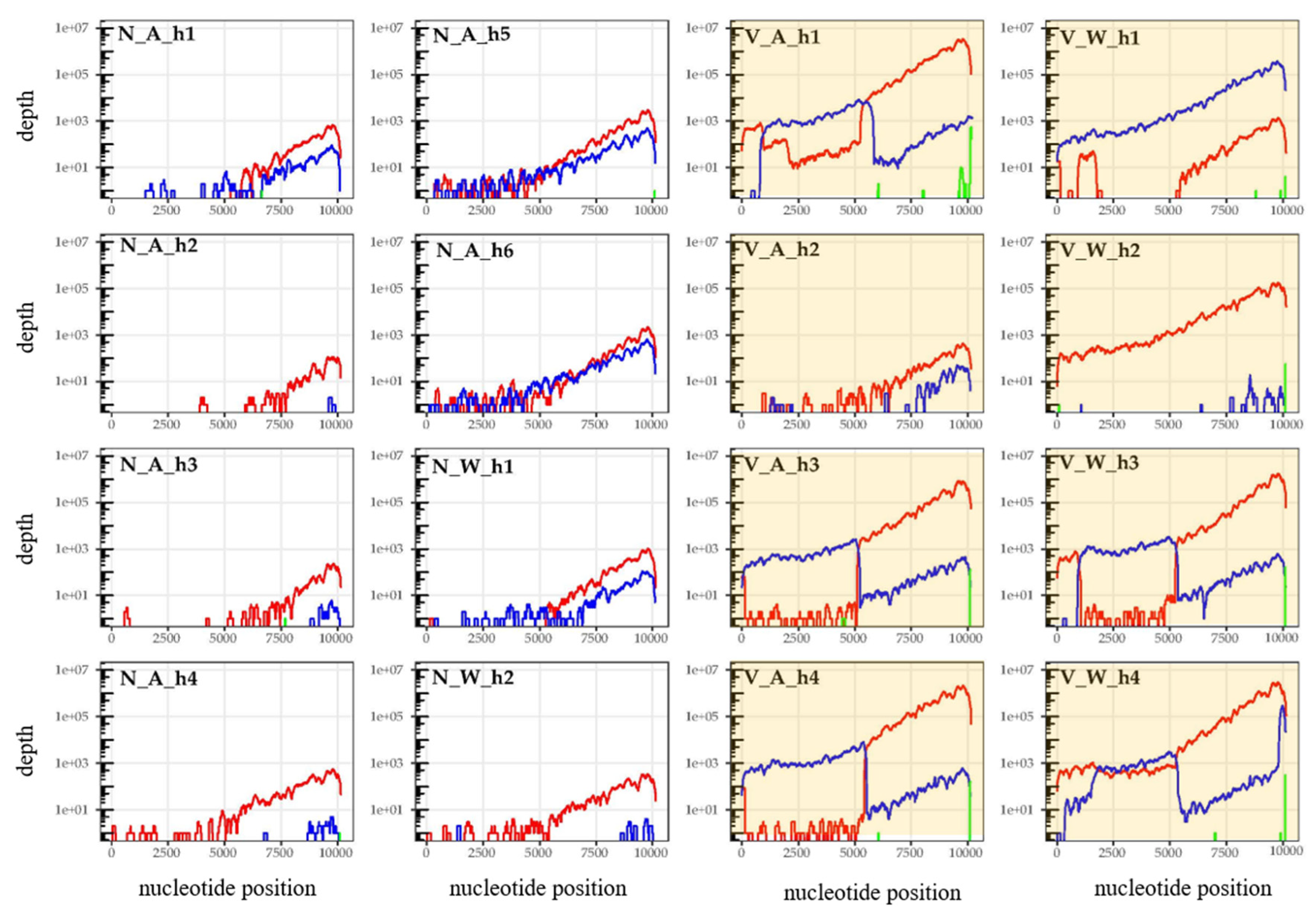
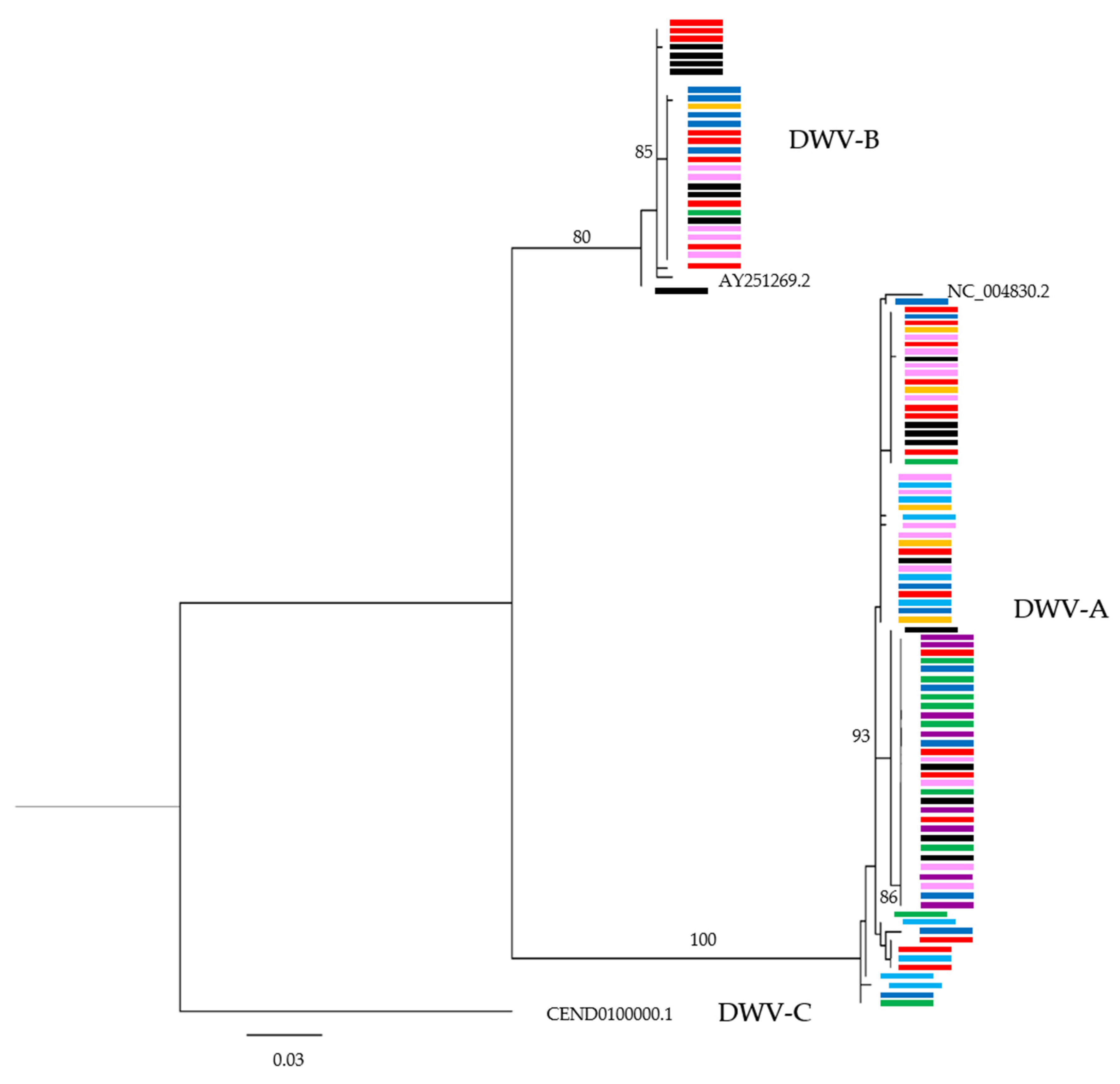
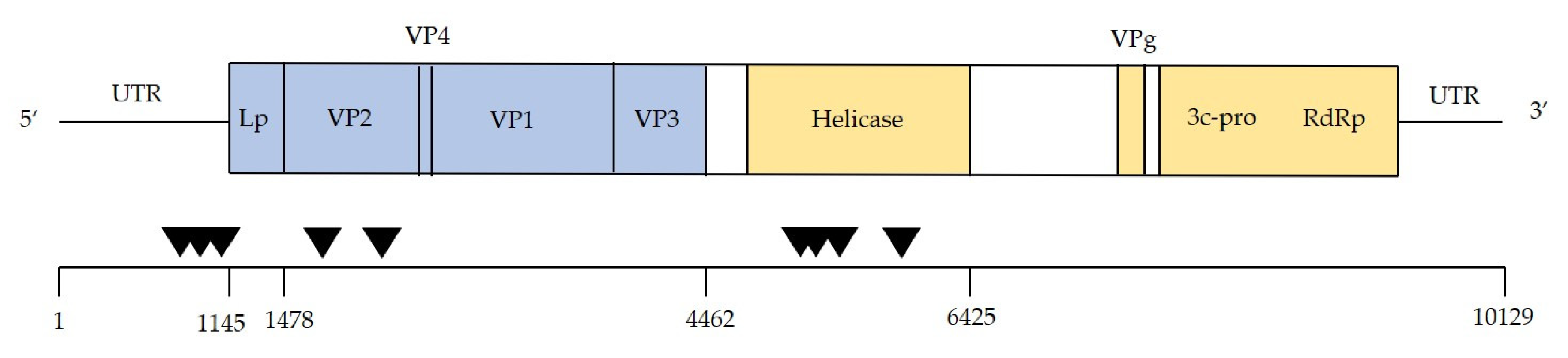
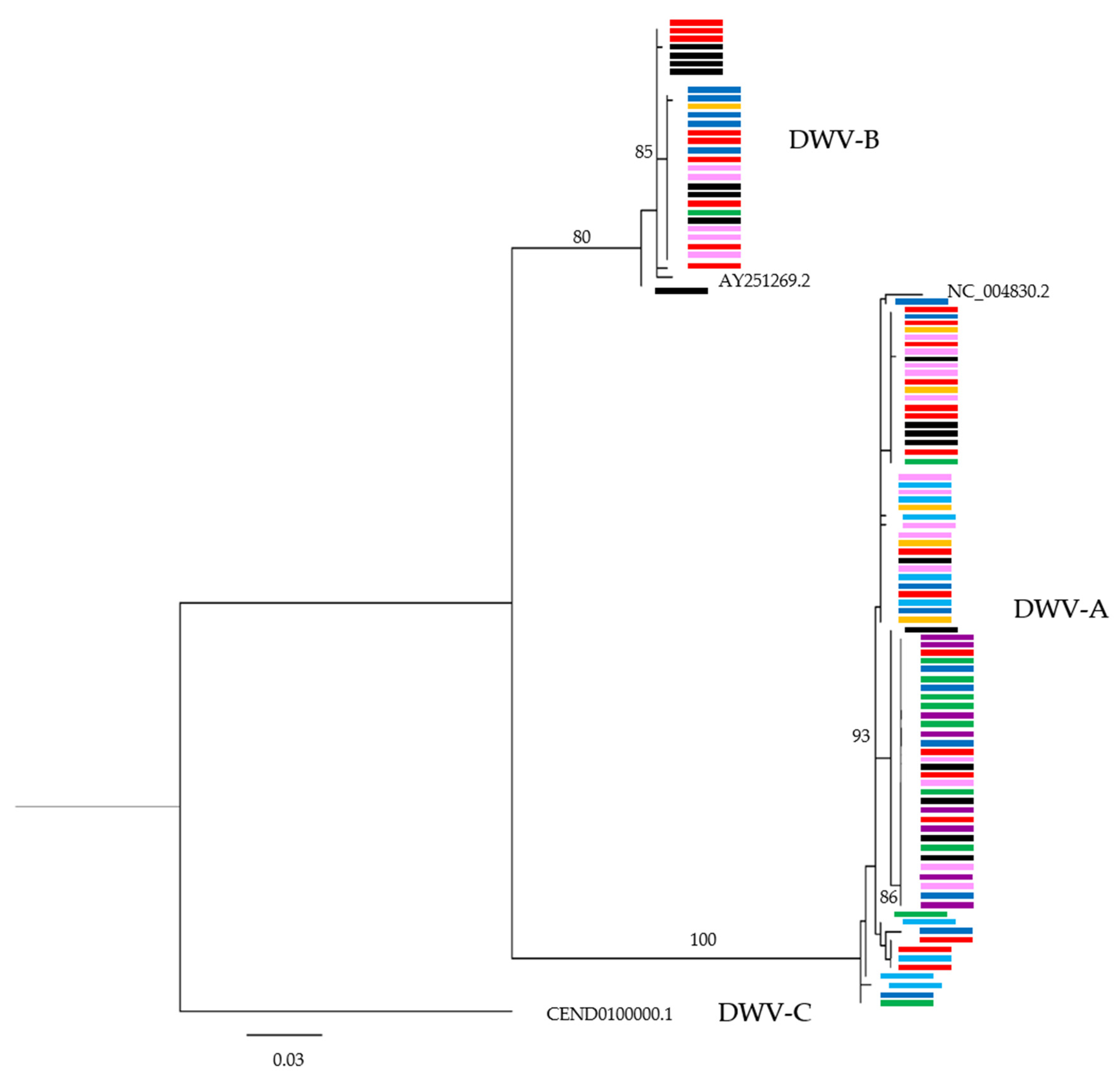
3.3. Are Particular DWV Variants or Recombinants Thereof Correlated with Host Taxon or Varroa Presence?
3.4. Are Common DWV Variants Found to Dominate Across Taxa?
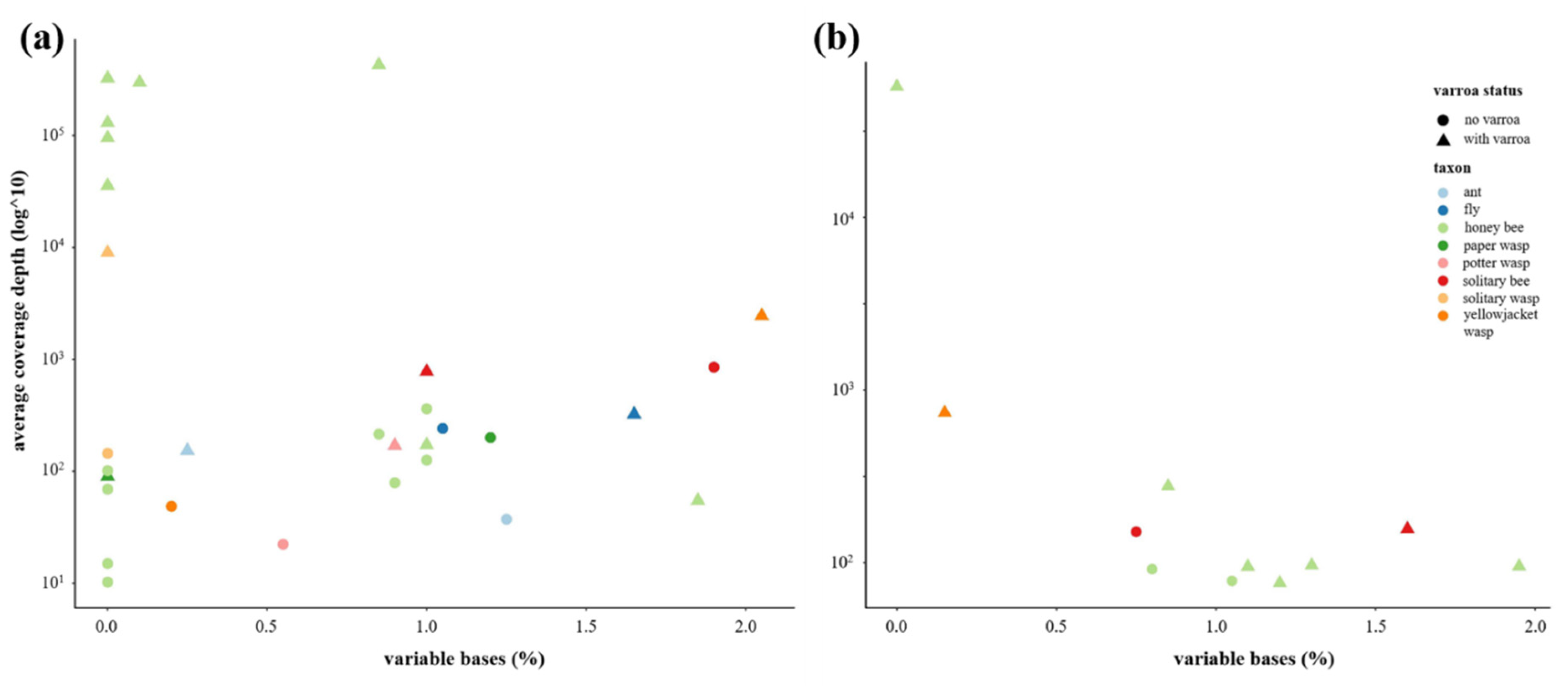
3.5. Has the Establishment of Varroa Caused a Decrease in DWV Quasispecies Diversity in Non-Apis Species?
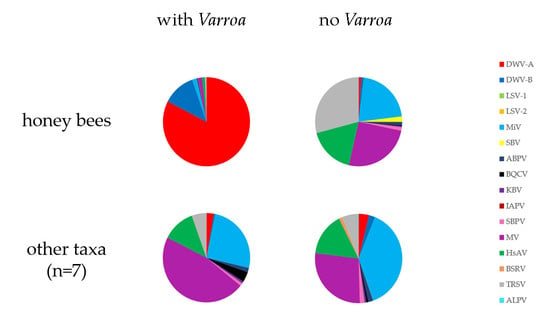
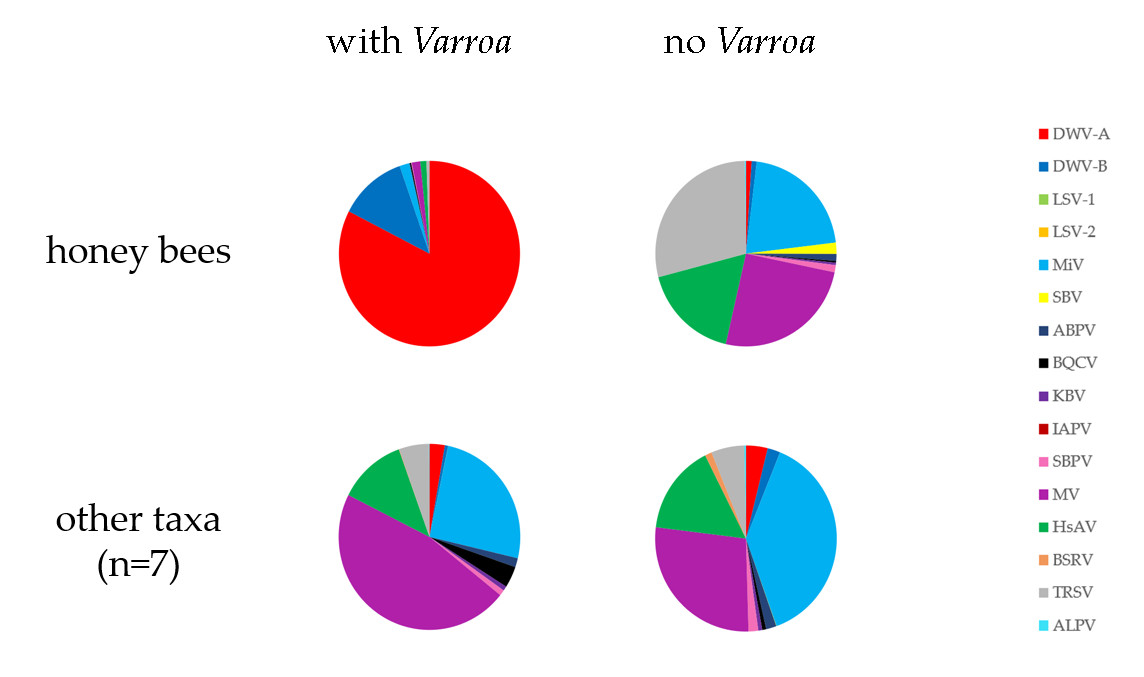
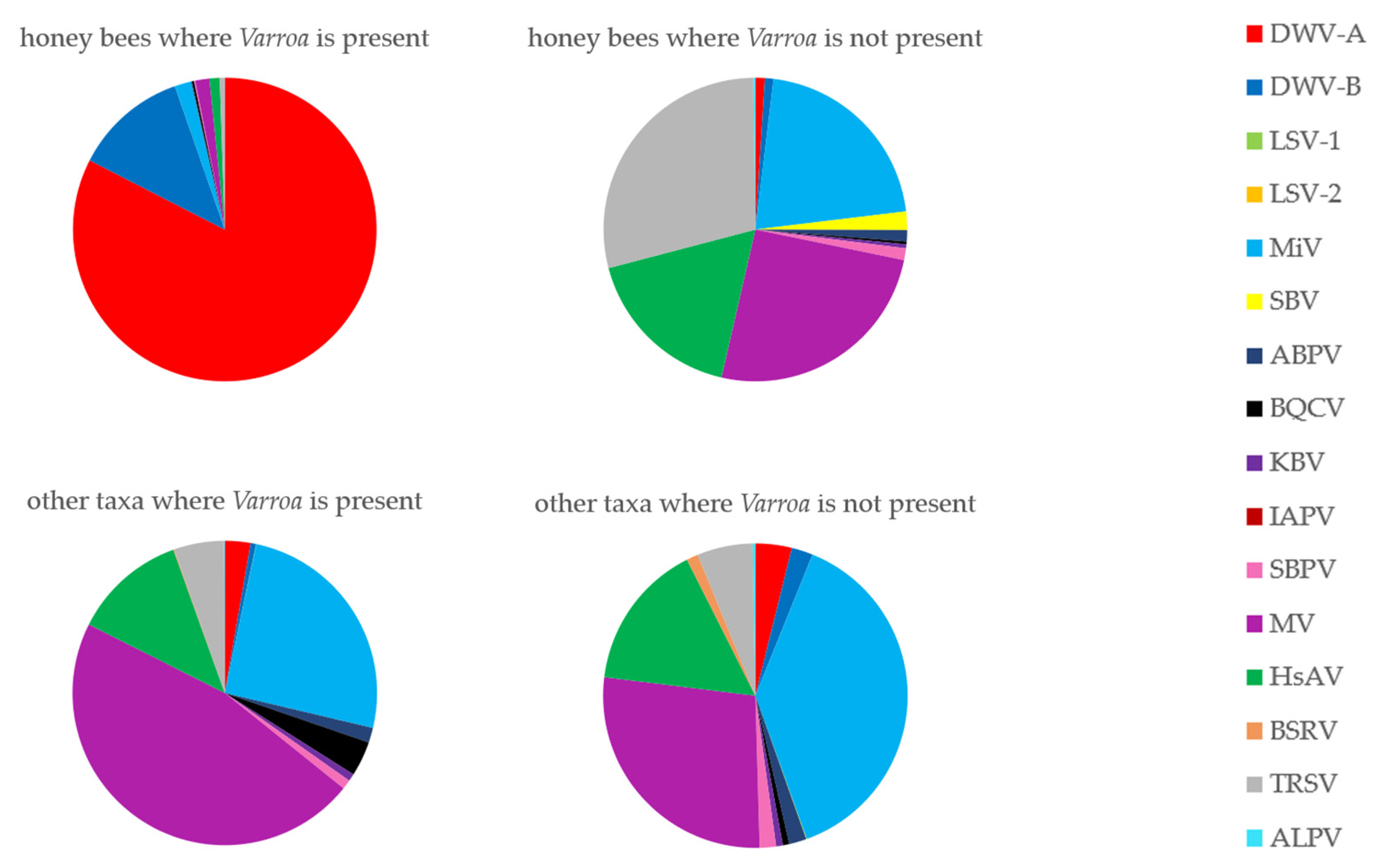
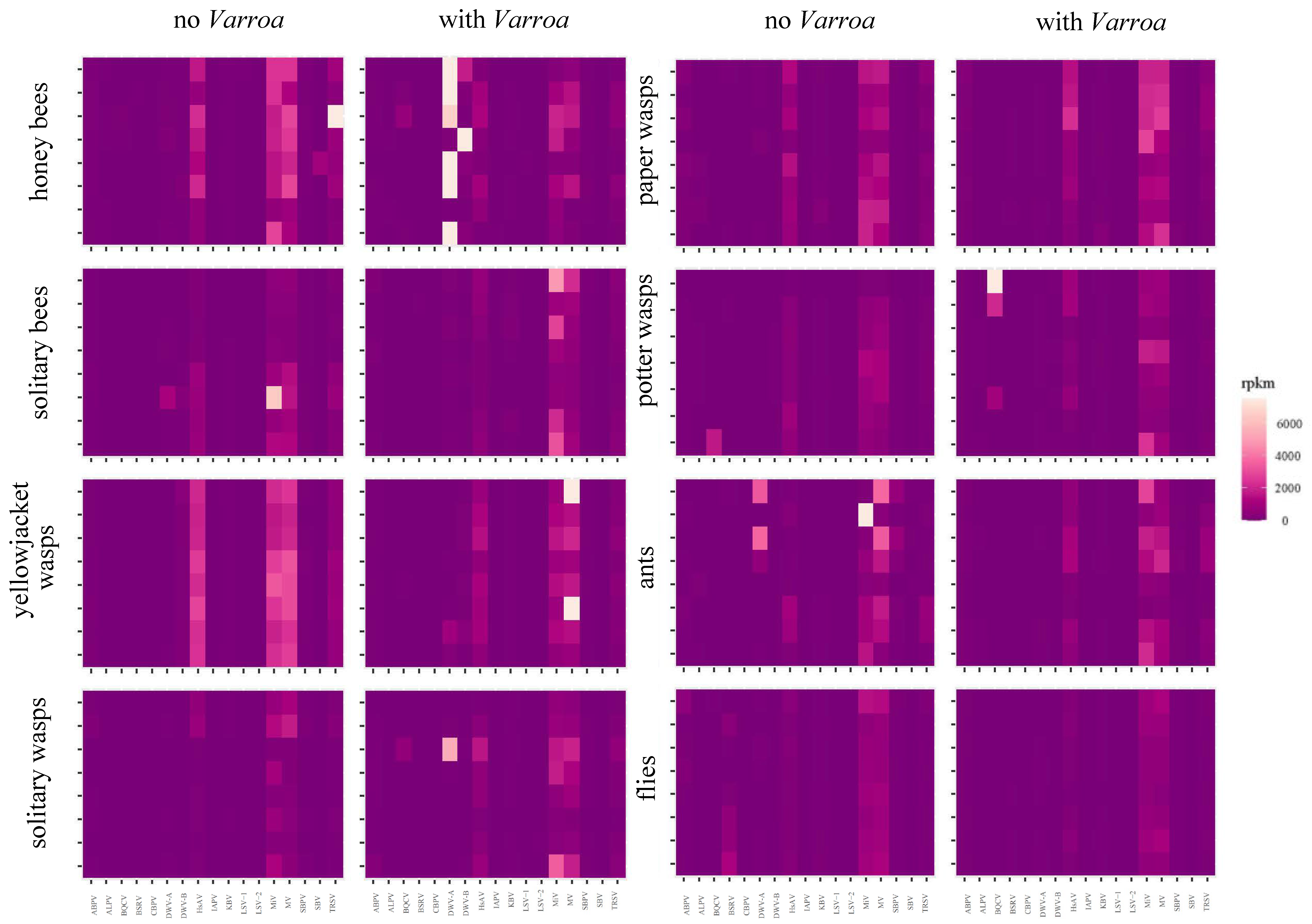
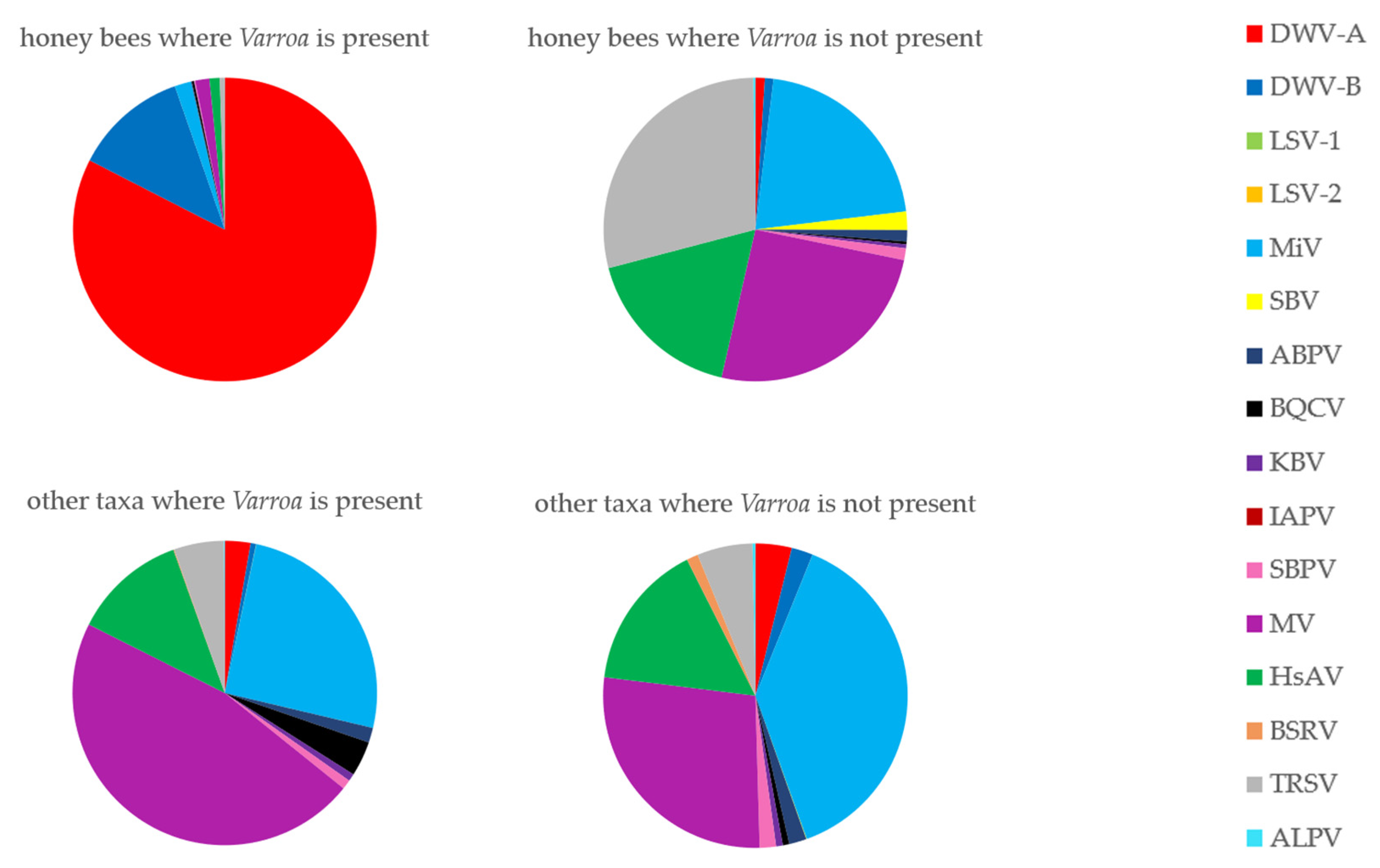
3.6. Has the Establishment of Varroa Caused a Shift in the Viral Landscapes of Insects of Different Taxa?
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oldroyd, B.P. Coevolution while you wait: Varroa jacobsoni, a new parasite of western honeybees. Trends Ecol. Evol. 1999, 14, 312–315. [Google Scholar] [CrossRef]
- Wilfert, L.; Long, G.; Leggett, H.C.; Schmid-Hempel, P.; Butlin, R.; Martin, S.J.M.; Boots, M. Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science 2016, 351, 594–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miranda, J.R.; Genersch, E. Deformed wing virus. J. Invertebr. Pathol. 2010, 103, S48–S61. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Highfield, A.C.; Brettell, L.; Villalobos, E.M.; Budge, G.E.; Powell, M.; Nikaido, S.; Schroeder, D.C. Global Honey Bee Viral Landscape Altered by a Parasitic Mite. Science 2012, 336, 1304–1306. [Google Scholar] [CrossRef]
- Ryabov, E.V.; Wood, G.R.; Fannon, J.M.; Moore, J.D.; Bull, J.C.; Chandler, D.; Mead, A.; Burroughs, N.; Evans, D.J. A Virulent Strain of Deformed Wing Virus (DWV) of Honeybees (Apis mellifera) Prevails after Varroa destructor-Mediated, or In Vitro, Transmission. PLoS Pathog. 2014, 10, e1004230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzi, G.; De Miranda, J.R.; Boniotti, M.B.; Cameron, C.E.; Lavazza, A.; Capucci, L.; Camazine, S.M.; Rossi, C. Molecular and Biological Characterization of Deformed Wing Virus of Honeybees (Apis mellifera L.). J. Virol. 2006, 80, 4998–5009. [Google Scholar] [CrossRef] [Green Version]
- Ongus, J.R.; Peters, D.; Bonmatin, J.-M.; Bengsch, E.; Vlak, J.M.; Van Oers, M.M. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J. Gen. Virol. 2004, 85, 3747–3755. [Google Scholar] [CrossRef]
- Mordecai, G.J.; Wilfert, L.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Diversity in a honey bee pathogen: First report of a third master variant of the Deformed Wing Virus quasispecies. ISME J. 2015, 10, 1264–1273. [Google Scholar] [CrossRef] [Green Version]
- Kevill, J.L.; De Souza, F.S.; Sharples, C.; Oliver, R.; Schroeder, D.C.; Martin, S.J. DWV-A Lethal to Honey Bees (Apis mellifera): A Colony Level Survey of DWV Variants (A, B, and C) in England, Wales, and 32 States across the US. Viruses 2019, 11, 426. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.J.; Brettell, L.E. Deformed Wing Virus in Honeybees and Other Insects. Annu. Rev. Virol. 2019, 6, 49–69. [Google Scholar] [CrossRef]
- Manley, R.; Temperton, B.; Doyle, T.; Gates, D.; Hedges, S.; Boots, M.; Wilfert, L. Knock-on community impacts of a novel vector: Spillover of emerging DWV-B from Varroa—Infested honeybees to wild bumblebees. Ecol. Lett. 2019, 22, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Murray, E.A.; Burand, J.; Trikoz, N.; Schnabel, J.; Grab, H.; Danforth, B.N. Viral transmission in honey bees and native bees, supported by a global black queen cell virus phylogeny. Environ. Microbiol. 2019, 21, 972–983. [Google Scholar] [CrossRef]
- Japoshvili, G.; Theodorou, P.; Husemann, M.; Japoshvili, G.; Kirkitadze, G.; Zhusupbaeva, A.; Paxton, R.J. Replication of honey bee-associated RNA viruses across multiple bee species in apple orchards of Georgia, Germany and Kyrgyzstan. J. Invertebr. Pathol. 2017, 146, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, J.; Villalobos, E.M.; Brettell, L.E.; Nikaido, S.; Graham, J.R.; Martin, S. Evidence of Varroa-mediated deformed wing virus spillover in Hawaii. J. Invertebr. Pathol. 2018, 151, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Furst, A.M.; McMahon, D.P.; Osborne, J.L.; Paxton, R.J.; Brown, M.J.F. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 2014, 506, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Tehel, A.; Brown, M.J.F.; Paxton, R. Impact of managed honey bee viruses on wild bees. Curr. Opin. Virol. 2016, 19, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettell, L.E.; Schroeder, D.C.; Martin, S.J. RNAseq Analysis Reveals Virus Diversity within Hawaiian Apiary Insect Communities. Viruses 2019, 11, 397. [Google Scholar] [CrossRef] [Green Version]
- Loope, K.J.; Baty, J.W.; Lester, P.J.; Rankin, E.E.W. Pathogen shifts in a honeybee predator following the arrival of the Varroa mite. Proc. R. Soc. B Biol. Sci. 2019, 286, 20182499. [Google Scholar] [CrossRef] [Green Version]
- Graystock, P.; Meeus, I.; Smagghe, G.; Goulson, D.; Hughes, W.O.H. The effects of single and mixed infections of Apicystis bombi and deformed wing virus in Bombus terrestris. Parasitology 2015, 143, 358–365. [Google Scholar] [CrossRef]
- Gusachenko, O.N.; Woodford, L.; Balbirnie-Cumming, K.; Ryabov, E.V.; Evans, D.J. Evidence for and against deformed wing virus spillover from honey bees to bumble bees: A reverse genetic analysis. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Zhang, X.; He, S.; Evans, J.D.; Pettis, J.; Yin, G.; Chen, Y. New evidence that deformed wing virus and black queen cell virus are multi-host pathogens. J. Invertebr. Pathol. 2012, 109, 156–159. [Google Scholar] [CrossRef]
- Ravoet, J.; De Smet, L.; Meeus, I.; Smagghe, G.; Wenseleers, T.; De Graaf, D.C. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 2014, 122, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; A Schmidt, H.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree-version 1.4. 4, a Graphical Viewer of Phylogenetic Trees. 2018, Computer Program Distributed by the Author. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 24 August 2020).
- McMahon, D.P.; Wilfert, L.; Paxton, R.J.; Brown, M.J. Emerging viruses in bees: From molecules to ecology. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2018; pp. 251–291. [Google Scholar] [CrossRef]
- Mordecai, G.J.; Brettell, L.E.; Pachori, P.; Villalobos, E.M.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Moku virus; a new Iflavirus found in wasps, honey bees and Varroa. Sci. Rep. 2016, 6, 34983. [Google Scholar] [CrossRef]
- Brettell, L.E.; Mordecai, G.J.; Pachori, P.; Martin, S.J. Novel RNA Virus Genome Discovered in Ghost Ants (Tapinoma melanocephalum) from Hawaii. Genome Announc. 2017, 5, e00669-17. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Usinglme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; Sage: Thousand Oaks, CA, USA, 2019. [Google Scholar]
- Lenth, R.V. Least-Squares Means: TheRPackagelsmeans. J. Stat. Softw. 2016, 69, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Mondet, F.; De Miranda, J.R.; Kretzschmar, A.; Le Conte, Y.; Mercer, A. On the Front Line: Quantitative Virus Dynamics in Honeybee (Apis mellifera L.) Colonies along a New Expansion Front of the Parasite Varroa destructor. PLoS Pathog. 2014, 10, e1004323. [Google Scholar] [CrossRef] [Green Version]
- Ryabov, E.V.; Childers, A.K.; Chen, Y.; Madella, S.; Nessa, A.; Vanengelsdorp, D.; Evans, J.D. Recent spread of Varroa destructor virus-1, a honey bee pathogen, in the United States. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- De Souza, F.S.; Allsopp, M.H.; Martin, S.J. Deformed wing virus prevalence and load in honeybees in South Africa. Arch. Virol 2020, accepted. [Google Scholar]
- Ryabov, E.V.; Childers, A.K.; Lopez, D.; Grubbs, K.; Posada-Florez, F.; Weaver, D.; Girten, W.; Vanengelsdorp, D.; Chen, Y.; Evans, J.D. Dynamic evolution in the key honey bee pathogen deformed wing virus: Novel insights into virulence and competition using reverse genetics. PLoS Biol. 2019, 17, e3000502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, A.M.; Remnant, E.J.; Buchmann, G.; Beekman, M. Accumulation and Competition amongst Deformed Wing Virus Genotypes in Naïve Australian Honeybees Provides Insight into the Increasing Global Prevalence of Genotype B. Front. Microbiol. 2020, 11, 620. [Google Scholar] [CrossRef]
- Tehel, A.; Vu, Q.; Bigot, D.; Gogol-Döring, A.; Koch, P.; Jenkins, C.; Doublet, V.; Theodorou, P.; Paxton, R. The Two Prevalent Genotypes of an Emerging Infectious Disease, Deformed Wing Virus, Cause Equally Low Pupal Mortality and Equally High Wing Deformities in Host Honey Bees. Viruses 2019, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Mordecai, G.J.; Brettell, L.E.; Martin, S.J.; Dixon, D.; Jones, I.M.; Schroeder, D.C. Superinfection exclusion and the long-term survival of honey bees in Varroa-infested colonies. ISME J. 2015, 10, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Dalmon, A.; Desbiez, C.; Coulon, M.; Thomasson, M.; Le Conte, Y.; Alaux, C.; Vallon, J.; Moury, B. Evidence for positive selection and recombination hotspots in Deformed wing virus (DWV). Sci. Rep. 2017, 7, srep41045. [Google Scholar] [CrossRef]
- Moore, J.; Jironkin, A.; Chandler, D.; Burroughs, N.; Evans, D.J.; Ryabov, E.V. Recombinants between Deformed wing virus and Varroa destructor virus-1 may prevail in Varroa destructor-infested honeybee colonies. J. Gen. Virol. 2010, 92, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Budge, G.E.; Watkins, M.; Bowman, A.S. Transcriptome analysis of the synganglion from the honey bee mite, Varroa destructor and RNAi knockdown of neural peptide targets. Insect Biochem. Mol. Biol. 2016, 70, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Gusachenko, O.N.; Woodford, L.; Balbirnie-Cumming, K.; Campbell, E.M.; Christie, C.R.; Bowman, A.S.; Evans, D.J. Green Bees: Reverse Genetic Analysis of Deformed Wing Virus Transmission, Replication, and Tropism. Viruses 2020, 12, 532. [Google Scholar] [CrossRef]
- Schläppi, D.; Lattrell, P.; Yañez, O.; Chejanovsky, N.; Neumann, P. Foodborne Transmission of Deformed Wing Virus to Ants (Myrmica rubra). Insects 2019, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Tapia-González, J.M.; Morfin, N.; Macías-Macías, J.O.; De La Mora, A.; Tapia-Rivera, J.C.; Ayala, R.; Contreras-Escareño, F.; Gashout, H.A.; Guzman-Novoa, E. Evidence of presence and replication of honey bee viruses among wild bee pollinators in subtropical environments. J. Invertebr. Pathol. 2019, 168, 107256. [Google Scholar] [CrossRef]
- Mazzei, M.; Carrozza, M.L.; Luisi, E.; Forzan, M.; Giusti, M.; Sagona, S.; Tolari, F.; Felicioli, A. Infectivity of DWV Associated to Flower Pollen: Experimental Evidence of a Horizontal Transmission Route. PLoS ONE 2014, 9, e113448. [Google Scholar] [CrossRef] [Green Version]
- Forzan, M.; Sagona, S.; Mazzei, M.; Felicioli, A. Detection of deformed wing virus in Vespa crabro. Bull. Insectol. 2017, 70, 261–265. [Google Scholar]
- Genersch, E.; Yue, C.; Fries, I.; De Miranda, J.R. Detection of Deformed wing virus, a honey bee viral pathogen, in bumble bees (Bombus terrestris and Bombus pascuorum) with wing deformities. J. Invertebr. Pathol. 2006, 91, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Dalmon, A.; Gayral, P.; Decante, D.; Klopp, C.; Bigot, D.; Thomasson, M.; Herniou, E.A.; Alaux, C.; Le Conte, Y. Viruses in the Invasive Hornet Vespa velutina. Viruses 2019, 11, 1041. [Google Scholar] [CrossRef] [Green Version]
- Tehel, A.; Streicher, T.; Tragust, S.; Paxton, R.J. Experimental infection of bumblebees with honeybee-associated viruses: No direct fitness costs but potential future threats to novel wild bee hosts. R. Soc. Open Sci. 2020, 7, 200480. [Google Scholar] [CrossRef]
- Dolezal, A.G.; Hendrix, S.D.; Scavo, N.A.; Carrillo-Tripp, J.; Harris, M.A.; Wheelock, M.J.; O’Neal, M.E.; Toth, A.L. Honey Bee Viruses in Wild Bees: Viral Prevalence, Loads, and Experimental Inoculation. PLoS ONE 2016, 11, e0166190. [Google Scholar] [CrossRef]
- Levitt, A.L.; Singh, R.; Cox-Foster, D.L.; Rajotte, E.; Hoover, K.; Ostiguy, N.; Holmes, E.C. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 2013, 176, 232–240. [Google Scholar] [CrossRef]
- Manley, R.; Boots, M.; Wilfert, L. REVIEW: Emerging viral disease risk to pollinating insects: Ecological, evolutionary and anthropogenic factors. J. Appl. Ecol. 2015, 52, 331–340. [Google Scholar] [CrossRef]
- Tentcheva, D.; Gauthier, L.; Zappulla, N.; Dainat, B.; Cousserans, F.; Colin, M.E.; Bergoin, M. Prevalence and Seasonal Variations of Six Bee Viruses in Apis mellifera L. and Varroa destructor Mite Populations in France. Appl. Environ. Microbiol. 2004, 70, 7185–7191. [Google Scholar] [CrossRef] [Green Version]
- Brettell, L.E.; Riegler, M.; O’Brien, C.; Cook, J.M. Occurrence of honey bee-associated pathogens in Varroa-free pollinator communities. J. Invertebr. Pathol. 2020, 171, 107344. [Google Scholar] [CrossRef]
- Fung, E. RNA Viruses in Australian Bees. Ph.D. Thesis, University of Adelaide, Adelaide, Australia, 2017. [Google Scholar]
- Roddy, K.M.; Arita-Tsutsumi, L. A History of honey bees in the Hawaiian Islands. J. Hawaii Pac. Agric. 1997, 8, 59–70. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brettell, L.E.; Schroeder, D.C.; Martin, S.J. RNAseq of Deformed Wing Virus and Other Honey Bee-Associated Viruses in Eight Insect Taxa with or without Varroa Infestation. Viruses 2020, 12, 1229. https://doi.org/10.3390/v12111229
Brettell LE, Schroeder DC, Martin SJ. RNAseq of Deformed Wing Virus and Other Honey Bee-Associated Viruses in Eight Insect Taxa with or without Varroa Infestation. Viruses. 2020; 12(11):1229. https://doi.org/10.3390/v12111229
Chicago/Turabian StyleBrettell, Laura E., Declan C. Schroeder, and Stephen J. Martin. 2020. "RNAseq of Deformed Wing Virus and Other Honey Bee-Associated Viruses in Eight Insect Taxa with or without Varroa Infestation" Viruses 12, no. 11: 1229. https://doi.org/10.3390/v12111229
APA StyleBrettell, L. E., Schroeder, D. C., & Martin, S. J. (2020). RNAseq of Deformed Wing Virus and Other Honey Bee-Associated Viruses in Eight Insect Taxa with or without Varroa Infestation. Viruses, 12(11), 1229. https://doi.org/10.3390/v12111229