Superimposition of Viral Protein Structures: A Means to Decipher the Phylogenies of Viruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structural Superimposition of Proteins and Homology of Viral Coat Proteins
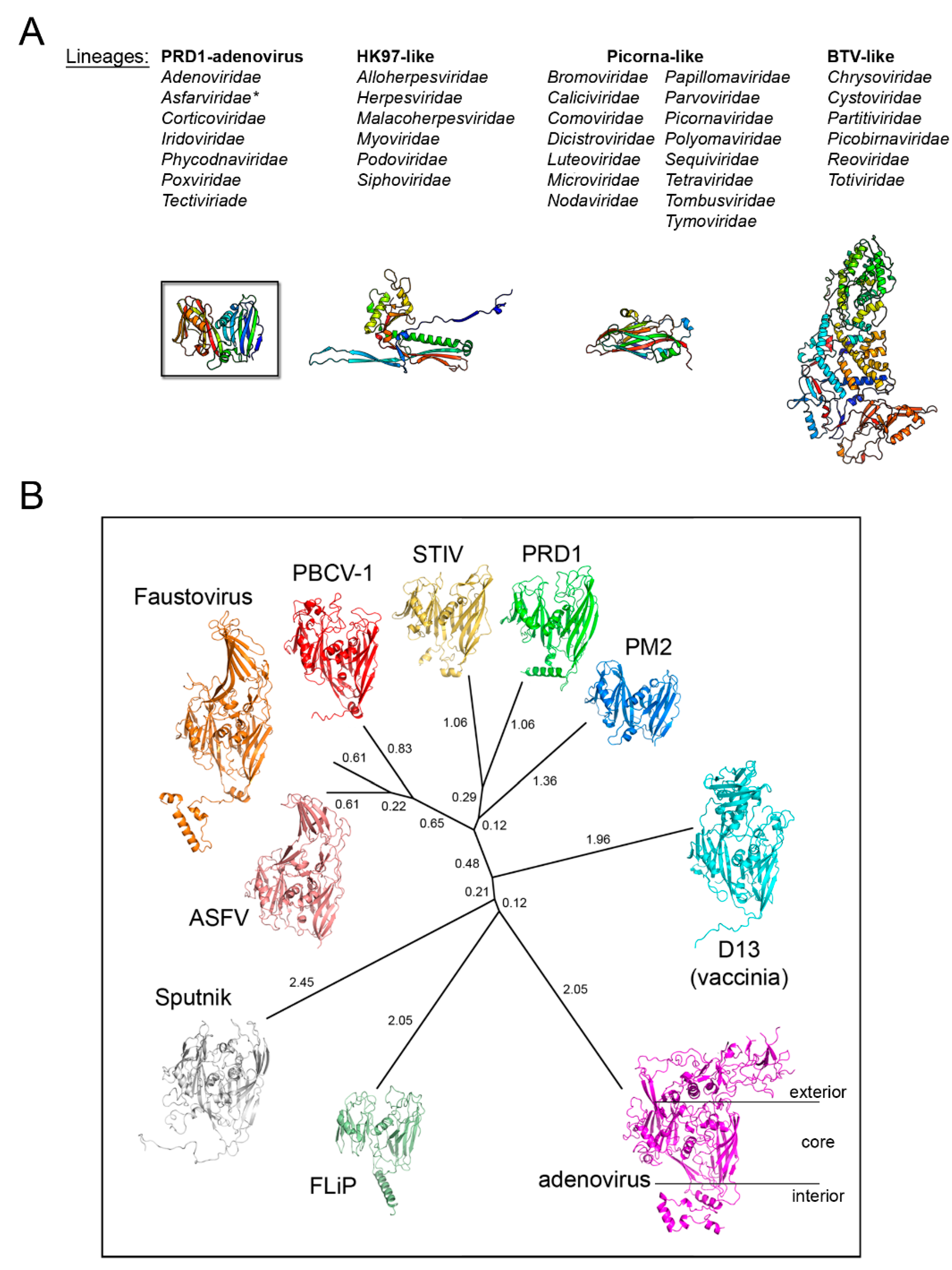
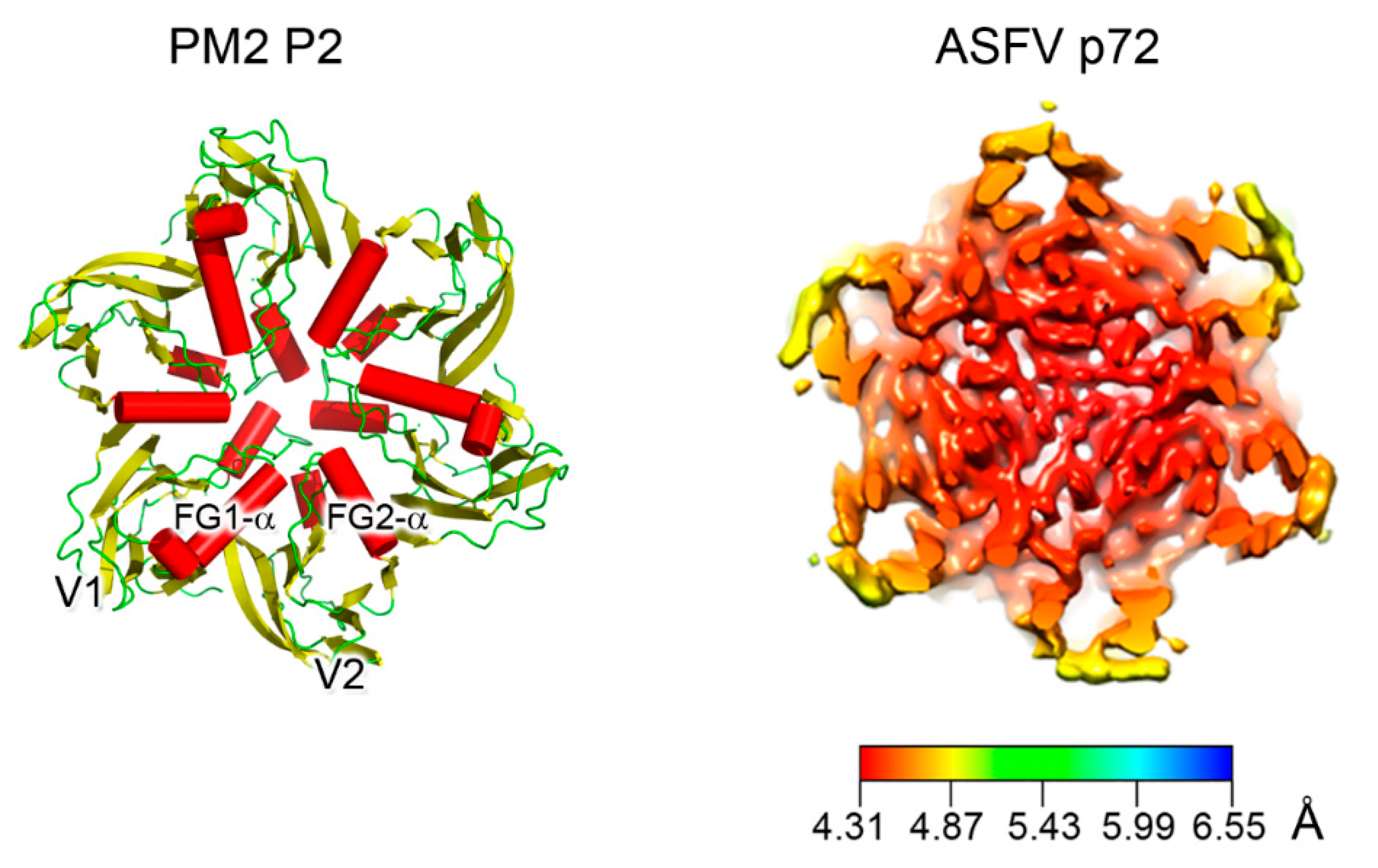
3. Major Capsid Proteins with a Double β-Barrel Fold: The Conceptualization of Viral Lineages
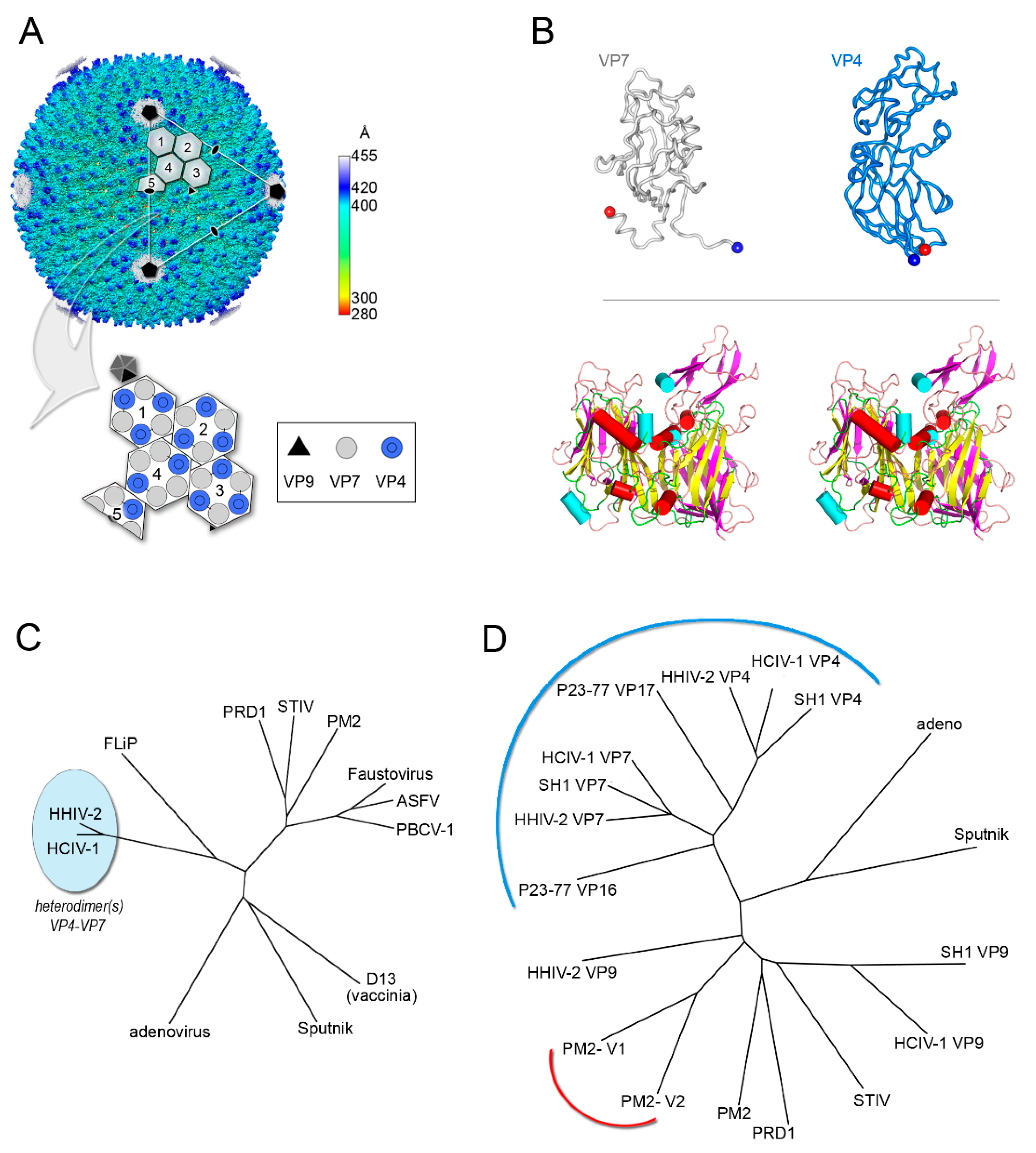
4. Viruses with Vertical Single β-barrel MCPs: A Clade of the PRD1-Adenovirus Lineage
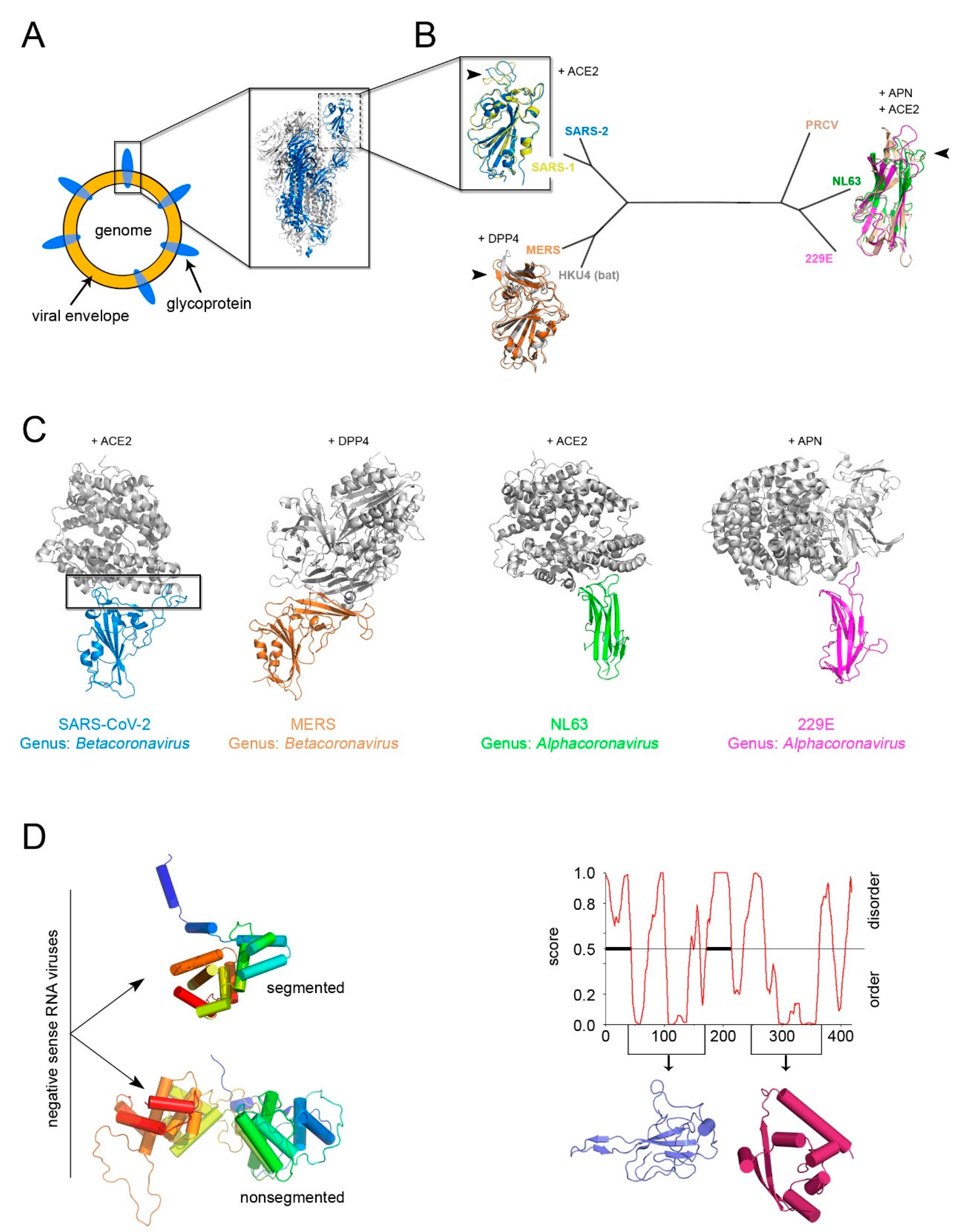
5. The Open Question of A Structure-Based Classification of Enveloped Viruses and the Example of Current SARS-CoV-2
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuan, R. Extremely high mutation rate of a hammerhead viroid. Science 2009, 323, 1308. [Google Scholar] [CrossRef]
- Koonin, E.V.; Galperin, M.Y. Evolutionary concept in genetics and genomics. In Sequence Evolution Function: Computational Approaches in Comparative Genomics; Kluwer Academic: Boston, MA, USA, 2003. [Google Scholar]
- Kimura, K.; Hunley, S.B.; Namy, L.L. Children’s use of comparison and function in novel object categorization. J. Exp. Child Psychol. 2018, 170, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Ravantti, J.; Abrescia, N.G. Classification of the viral world based on atomic level structures. In Encyclopedia of Virology; Bamford, D., Zuckerman, M., Eds.; Academic Press-Elsevier: Amsterdam, The Netherlands, 2021; In Press. [Google Scholar]
- Abrescia, N. Evolution steered by structure. In Encyclopedia of Virology; Bamford, D., Zuckerman, M., Eds.; Academic Press-Elsevier: Amsterdam, The Netherlands, 2021; In Press. [Google Scholar]
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.I.; Subramaniam, S.; Abrescia, N.G. The democratization of cryo-EM. Nat. Methods 2016, 13, 607–608. [Google Scholar] [CrossRef]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A.C. Structure of haemoglobin: A three-dimensional Fourier synthesis at 5.5-Å. resolution, obtained by X-ray analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F.; Muirhead, H.; Cox, J.M.; Goaman, L.C. Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 Å resolution: The atomic model. Nature 1968, 219, 131–139. [Google Scholar] [CrossRef]
- Huber, R.; Epp, O.; Steigemann, W.; Formanek, H. The atomic structure of erythrocruorin in the light of the chemical sequence and its comparison with myoglobin. Eur. J. Biochem. 1971, 19, 42–50. [Google Scholar] [CrossRef]
- Rao, S.T.; Rossmann, M.G. Comparison of super-secondary structures in proteins. J. Mol. Biol. 1973, 76, 241–256. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Argos, P. Exploring structural homology of proteins. J. Mol. Biol. 1976, 105, 75–95. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Definition of general topological equivalence in protein structures. A procedure involving comparison of properties and relationships through simulated annealing and dynamic programming. J. Mol. Biol. 1990, 212, 403–428. [Google Scholar]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993, 233, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.I.; Levine, M.; Muirhead, H.; Stammers, D.K. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 Å. J. Mol. Biol. 1979, 134, 109–142. [Google Scholar] [CrossRef]
- Ravantti, J.; Bamford, D.; Stuart, D.I. Automatic comparison and classification of protein structures. J. Struct. Biol. 2013, 183, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C.; Olson, A.J.; Schutt, C.E.; Winkler, F.K.; Bricogne, G. Tomato bushy stunt virus at 2.9 Å resolution. Nature 1978, 276, 368–373. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Abdel-Meguid, S.S.; Johnson, J.E.; Leslie, A.G.; Rayment, I.; Rossmann, M.G.; Suck, D.; Tsukihara, T. Structure of southern bean mosaic virus at 2.8 Å resolution. Nature 1980, 286, 33–39. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar] [CrossRef]
- Hogle, J.M.; Chow, M.; Filman, D.J. Three-dimensional structure of poliovirus at 2.9 Å resolution. Science 1985, 229, 1358–1365. [Google Scholar] [CrossRef]
- Burnett, R.M.; Athappilly, F.K.; Cai, Z.; Furcinitti, P.S.; Korn, A.P.; Murali, R.; van Oostrum, J. Structure of the Adenovirus Virion. In Use of X-ray Crystallography in the Design of Antiviral Agents; Academic Press Inc.: San Diego, CA, USA, 1990. [Google Scholar]
- Abrescia, N.G.; Cockburn, J.J.B.; Grimes, J.M.; Sutton, G.C.; Diprose, J.M.; Butcher, S.J.; Fuller, S.D.; San Martín, C.; Burnett, R.M.; Stuart, D.I.; et al. Insights into assembly from structural analysis of bacteriophage PRD1. Nature 2004, 432, 68–74. [Google Scholar]
- Cockburn, J.J.; Abrescia, N.G.; Grimes, J.M.; Sutton, G.C.; Diprose, J.M.; Benevides, J.M.; Thomas, G.J., Jr.; Bamford, J.K.H.; Bamford, D.H.; Stuart, D.I. Membrane structure and interactions with protein and DNA in bacteriophage PRD1. Nature 2004, 432, 122–125. [Google Scholar] [CrossRef]
- Chapman, M.S.; Liljas, L. Structural folds of viral proteins. Adv. Protein Chem. 2003, 64, 125–196. [Google Scholar]
- Benson, S.D.; Bamford, J.K.; Bamford, D.H.; Burnett, R.M. Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures. Cell 1999, 98, 825–833. [Google Scholar] [CrossRef]
- Bamford, D.H.; Burnett, R.M.; Stuart, D.I. Evolution of viral structure. Theor. Popul. Biol. 2002, 61, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Nandhagopal, N.; Simpson, A.A.; Gurnon, J.R.; Yan, X.; Baker, T.S.; Graves, M.V.; Van Etten, J.L.; Rossmann, M.G. The structure and evolution of the major capsid protein of a large, lipid-containing DNA virus. Proc. Natl. Acad. Sci. USA 2002, 99, 14758–14763. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, N.G.; Grimes, J.M.; Kivelä, H.M.; Assenberg, R.; Sutton, G.C.; Butcher, S.J.; Bamford, J.K.H.; Bamford, D.H.; Stuart, D.I. Insights into virus evolution and membrane biogenesis from the structure of the marine lipid-containing bacteriophage PM2. Mol. Cell 2008, 31, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Leiman, P.G.; Shneider, M.M.; Ahvazi, B.; Boeshans, K.M.; Steven, A.C.; Black, L.W.; Mesyanzhinov, V.V.; Rossmann, M.G. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. USA 2005, 102, 7163–7168. [Google Scholar] [CrossRef] [PubMed]
- Pietila, M.K.; Laurinmäki, P.; Russell, D.A.; Ko, C.-C.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc. Natl. Acad. Sci. USA 2013, 110, 10604–10609. [Google Scholar] [CrossRef]
- Bahar, M.W.; Graham, S.C.; Stuart, D.I.; Grimes, J.M. Insights into the evolution of a complex virus from the crystal structure of vaccinia virus d13. Structure 2011, 19, 1011–1020. [Google Scholar] [CrossRef]
- Klose, T.; Reteno, D.G.; Benamar, S.; Hollerbach, A.; Colson, P.; La Scola, B.; Rossmann, M.G. Structure of faustovirus, a large dsDNA virus. Proc. Natl. Acad. Sci. USA 2016, 113, 6206–6211. [Google Scholar] [CrossRef]
- Khayat, R.; Tang, L.; Larson, E.T.; Lawrence, C.M.; Young, M.; Johnson, J.E. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc. Natl. Acad. Sci. USA 2005, 102, 18944–18949. [Google Scholar] [CrossRef]
- Hyun, J.K.; Accurso, C.; Hijnen, M.; Schult, P.; Pettikiriarachchi, A.; Mitra, A.K.; Coulibaly, F. Membrane remodeling by the double-barrel scaffolding protein of poxvirus. PLoS Pathog. 2011, 7, e1002239. [Google Scholar] [CrossRef]
- Duda, R.L.; Teschke, C.M. The amazingHK97 fold: Versatile results of modest differences. Curr. Opin. Virol. 2019, 36, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, G. Fry, Ravantti, Bamford, Stuart, What does it take to make a virus: The concept of the viral “self”. In Emerging Topics in Physical Virology; Twarock, P.G.S.R., Ed.; Imperial College Press: London, UK, 2010; pp. 35–58. [Google Scholar]
- Abrescia, N.G.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure Unifies the Viral Universe. Annu. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient Virus World and evolution of cells. Biol. Direct. 2006, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origin of viruses: Primordial replicators recruiting capsids from hosts. Nat. Rev. Microbiol. 2019, 17, 449–458. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses Executive Committee. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef]
- Laanto, E.; Mäntynen, S.; De Colibus, L.; Marjakangas, J.; Gillum, A.; Stuart, D.I.; Ravantti, J.J.; Huiskonen, J.T.; Sundberg, L.R. Virus found in a boreal lake links ssDNA and dsDNA viruses. Proc. Natl. Acad. Sci. USA 2017, 114, 8378–8383. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, S.; Xiang, Y.; Wong, J.; Klose, T.; Raoult, D.; Rossmann, M.G. Structure of Sputnik, a virophage, at 3.5-A resolution. Proc. Natl. Acad. Sci. USA 2012, 109, 18431–18436. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Polintons: A hotbed of eukaryotic virus, transposon and plasmid evolution. Nat. Rev. Microbiol. 2015, 13, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Caspar, D.L.; Klug, A. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.A.; Nandhagopal, N.; Van Etten, J.L.; Rossmann, M.G. Structural analyses of Phycodnaviridae and Iridoviridae. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Andres, G.; Charro, D.; Matamoros, T.; Dillard, R.S.; Abrescia, N.G. The cryo-EM structure of African swine fever virus unravels a unique architecture comprising two icosahedral protein capsids and two lipoprotein membranes. J. Biol. Chem. 2020, 295, 1–12. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, B.; Qian, N.; Zhang, F.; Tan, X.; Lei, J.; Xiang, Y. Structure of the African swine fever virus major capsid protein p72. Cell Res. 2019, 29, 953–955. [Google Scholar] [CrossRef]
- Liu, S.; Luo, Y.; Wang, Y.; Li, S.; Zhao, Z.; Bi, Y.; Sun, J.; Peng, R.; Song, H.; Zhu, D.; et al. Cryo-EM Structure of the African Swine Fever Virus. Cell Host Microbe 2019, 26, 836–843.e3. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef]
- Fang, Q.; Zhu, D.; Agarkova, I.; Adhikari, J.; Klose, T.; Liu, Y.; Chen, Z.; Sun, Y.; Gross, M.L.; Van Etten, J.L.; et al. Near-atomic structure of a giant virus. Nat. Commun. 2019, 10, 388. [Google Scholar] [CrossRef]
- Born, D.; Reuter, L.; Mersdorf, U.; Mueller, M.; Fischer, M.G.; Meinhart, A.; Reinstein, J. Capsid protein structure, self-assembly, and processing reveal morphogenesis of the marine virophage mavirus. Proc. Natl. Acad. Sci. USA 2018, 115, 7332–7337. [Google Scholar] [CrossRef] [PubMed]
- Rux, J.J.; Burnett, R.M. Type-specific epitope locations revealed by X-ray crystallographic study of adenovirus type 5 hexon. Mol. Ther. 2000, 1, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Ernst, P.; Honegger, A.; Suomalainen, M.; Zimmermann, M.; Braun, L.; Stauffer, S.; Thom, C.; Dreier, B.; Eibauer, M.; et al. Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat. Commun. 2018, 9, 450. [Google Scholar] [CrossRef] [PubMed]
- Prill, J.M.; Espenlaub, S.; Samen, U.; Engler, T.; Schmidt, E.; Vetrini, F.; Rosewell, A.; Grove, N.; Palmer, D.; Ng, P.; et al. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from Kupffer cells. Mol. Ther. 2011, 19, 83–92. [Google Scholar] [CrossRef]
- Kauffman, K.M.; Hussain, F.A.; Yang, J.; Arevalo, P.; Brown, J.M.; Chang, W.K.; VanInsberghe, D.; Elsherbini, J.; Sharma, R.S.; Cutler, M.B.; et al. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature 2018, 554, 118–122. [Google Scholar] [CrossRef]
- Porter, K.; Kukkaro, P.; Bamford, J.K.; Bath, C.; Kivelä, H.M.; Dyall-Smith, M.L.; Bamford, D.H. SH1: A novel, spherical halovirus isolated from an Australian hypersaline lake. Virology 2005, 335, 22–33. [Google Scholar] [CrossRef]
- Kivela, H.M.; Roine, E.; Kukkaro, P.; Laurinavicius, S.; Somerharju, P.; Bamford, D.H. Quantitative dissociation of archaeal virus SH1 reveals distinct capsid proteins and a lipid core. Virology 2006, 356, 4–11. [Google Scholar] [CrossRef]
- Rissanen, I.; Pawlowski, A.; Harlos, K.; Grimes, J.M.; Stuart, D.I.; Bamford, J.K.H. Crystallization and preliminary crystallographic analysis of the major capsid proteins VP16 and VP17 of bacteriophage P23-77. Acta Crystallogr. 2012, 68, 580–583. [Google Scholar] [CrossRef]
- Gil-Carton, D.; Jaakkola, S.T.; Charro, D.; Peralta, B.; Castaño-Díez, D.; Oksanen, H.M.; Bamford, D.H.; Abrescia, N.G. Insight into the Assembly of Viruses with Vertical Single beta-barrel Major Capsid Proteins. Structure 2015, 23, 1866–1877. [Google Scholar] [CrossRef]
- Pawlowski, A.; Rissanen, I.; Bamford, J.K.; Krupovic, M.; Jalasvuori, M. Gammasphaerolipovirus, a newly proposed bacteriophage genus, unifies viruses of halophilic archaea and thermophilic bacteria within the novel family Sphaerolipoviridae. Arch. Virol. 2014, 159, 1541–1554. [Google Scholar] [CrossRef]
- Santos-Pérez, I.; Charro, D.; Gil-Carton, D.; Azkargorta, M.; Elortza, F.; Bamford, D.H.; Oksanen, H.M.; Abrescia, N.G. Structural basis for assembly of vertical single beta-barrel viruses. Nat. Commun. 2019, 10, 1184. [Google Scholar] [CrossRef] [PubMed]
- De Colibus, L.; Roine, E.; Walter, T.S.; Ilca, S.L.; Wang, X.; Wang, N.; Roseman, A.M.; Bamford, D.H.; Huiskonen, J.T.; Stuart, D.I. Assembly of complex viruses exemplified by a halophilic euryarchaeal virus. Nat. Commun. 2019, 10, 1456. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Vargas, J.; Krey, T.; Valansi, C.; Avinoam, O.; Haouz, A.; Jamin, M.; Raveh-Barak, H.; Podbilewicz, B.; Rey, F.A. Structural basis of eukaryotic cell-cell fusion. Cell 2014, 157, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.M.; Stelfox, A.J.; Bowden, T.A. Unraveling virus relationships by structure-based phylogenetic classification. Virus Evol. 2020, 6, veaa003. [Google Scholar] [CrossRef]
- Ye, Q.; West, A.M.V.; Silletti, S.; Corbett, K.D. Architecture and self-assembly of the SARS-CoV-2 nucleocapsid protein. Protein Sci. 2020, 29, 1890–1901. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravantti, J.J.; Martinez-Castillo, A.; Abrescia, N.G.A. Superimposition of Viral Protein Structures: A Means to Decipher the Phylogenies of Viruses. Viruses 2020, 12, 1146. https://doi.org/10.3390/v12101146
Ravantti JJ, Martinez-Castillo A, Abrescia NGA. Superimposition of Viral Protein Structures: A Means to Decipher the Phylogenies of Viruses. Viruses. 2020; 12(10):1146. https://doi.org/10.3390/v12101146
Chicago/Turabian StyleRavantti, Janne J., Ane Martinez-Castillo, and Nicola G.A. Abrescia. 2020. "Superimposition of Viral Protein Structures: A Means to Decipher the Phylogenies of Viruses" Viruses 12, no. 10: 1146. https://doi.org/10.3390/v12101146
APA StyleRavantti, J. J., Martinez-Castillo, A., & Abrescia, N. G. A. (2020). Superimposition of Viral Protein Structures: A Means to Decipher the Phylogenies of Viruses. Viruses, 12(10), 1146. https://doi.org/10.3390/v12101146