Characterization of Emerging Swine Viral Diseases through Oxford Nanopore Sequencing Using Senecavirus A as a Model



Abstract
1. Introduction
2. Materials and Methods
2.1. SVA Samples
2.2. RNA Extraction
2.3. Oxford Nanopore Sequencing
2.4. Bioinformatics Analysis
2.5. SVA Consensus Generation and Optimization
2.6. Sanger Sequencing and Analysis
2.7. Analytical Sensitivity Determination
2.8. Oxford Nanopore MinION Sequencing Data and Analysis Pipelines
3. Results
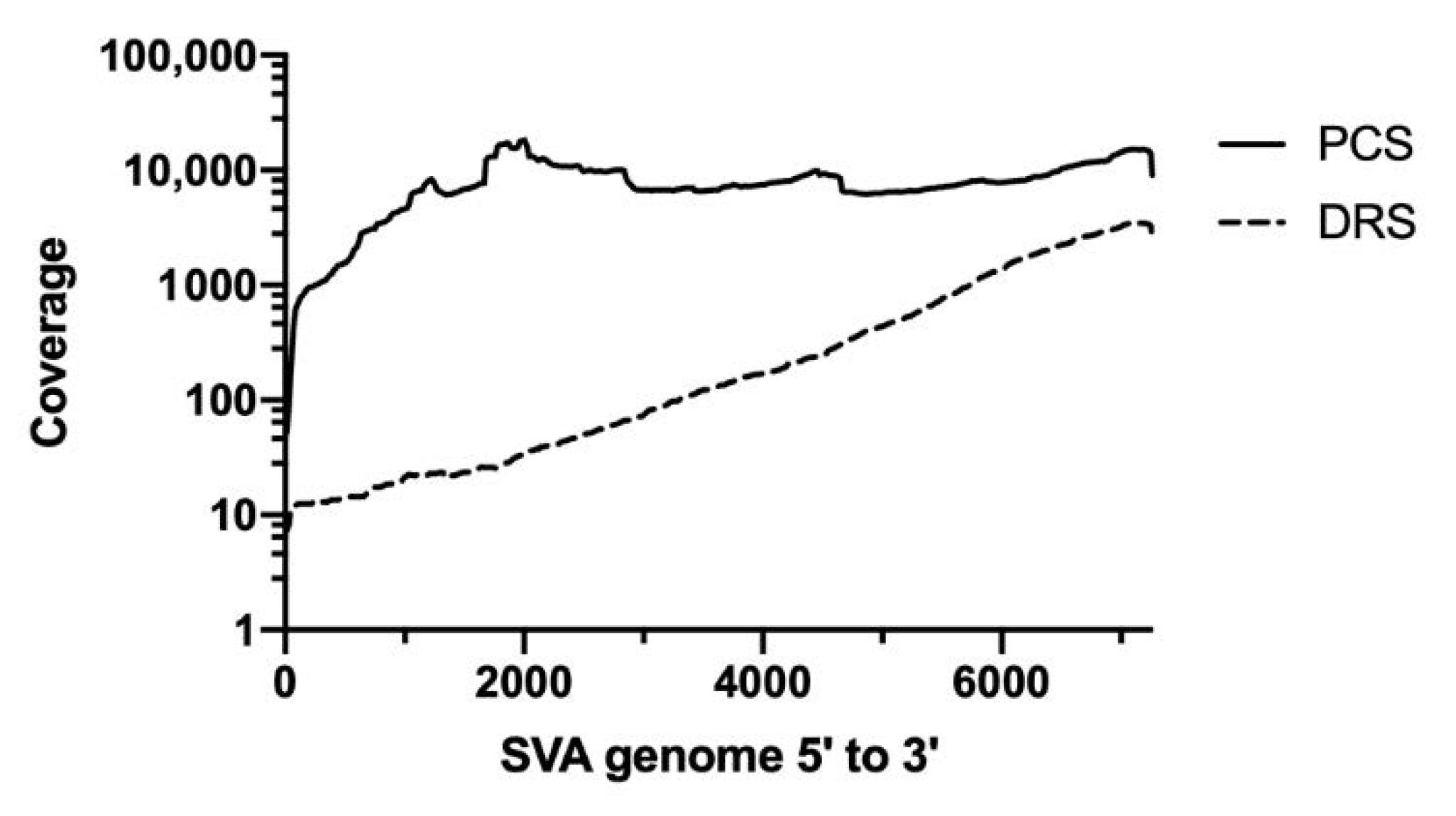
3.1. Assessment of Raw Reads from Direct RNA Sequencing and PCR-cDNA Sequencing
3.2. Optimization of Consensus Sequence Generation
3.3. Determination of Analytical Sensitivity of Direct RNA and PCR-cDNA Sequencing
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meng, X.J. Emerging and Re-Emerging Swine Viruses. Transbound. Emerg. Dis. 2012, 59 (Suppl. 1), 85–102. [Google Scholar] [CrossRef]
- Devaux, C.A. Emerging and re-emerging viruses: A global challenge illustrated by Chikungunya virus outbreaks. World J. Virol. 2012, 1, 11–22. [Google Scholar] [CrossRef]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Chapter 15-Emerging Virus Diseases. In Fenner and White’s Medical Virology, 5th ed.; Burrell, C.J., Howard, C.R., Murphy, F.A., Eds.; Academic Press: London, UK, 2017; pp. 217–225. [Google Scholar]
- Jones, G.; Patel, N.; Levy, M.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Lunney, J.K.; Benfield, D.A.; Rowland, R.R. Porcine reproductive and respiratory syndrome virus: An update on an emerging and re-emerging viral disease of swine. Virus Res. 2010, 154, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J.; Kekarainen, T.; Cortey, M. The natural history of porcine circovirus type 2: From an inoffensive virus to a devastating swine disease? Veter Microbiol. 2013, 165, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Goede, D.; Morrison, R.B. Production impact & time to stability in sow herds infected with porcine epidemic diarrhea virus (PEDV). Prev. Veter Med. 2016, 123, 202–207. [Google Scholar]
- Jurado, C.; Mur, L.; Aguirreburualde, M.S.P.; Cadenas-Fernández, E.; Martínez-López, B.; Sánchez-Vizcaíno, J.M.; Perez, A. Risk of African swine fever virus introduction into the United States through smuggling of pork in air passenger luggage. Sci. Rep. 2019, 9, 14423. [Google Scholar] [CrossRef] [PubMed]
- Hales, L.M.; Knowles, N.J.; Reddy, P.S.; Xu, L.; Hay, C.; Hallenbeck, P.L. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J. Gen. Virol. 2008, 89, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, S.; Reddy, S.P.; Loo, J.; Idamakanti, N.; Hallenbeck, P.L.; Reddy, V.S. Structure of Seneca Valley Virus-001: An oncolytic picornavirus representing a new genus. Structure 2008, 16, 1555–1561. [Google Scholar] [CrossRef]
- Burke, M.J. Oncolytic Seneca Valley Virus: Past perspectives and future directions. Oncolytic Virotherapy 2016, 5, 81–89. [Google Scholar] [CrossRef]
- Leme, R.D.A.; Zotti, E.; Alcântara, B.K.; Oliveira, M.V.; Freitas, L.A.; Alfieri, A.F.; Alfieri, A.A. Senecavirus A: An Emerging Vesicular Infection in Brazilian Pig Herds. Transbound. Emerg. Dis. 2015, 62, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhao, X.; Chen, Y.; He, X.; Zhang, G.; Ma, J. Complete Genome Sequence of Seneca Valley Virus CH-01-2015 Identified in China. Genome Announc. 2016, 4, e01509-15. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Canon, A.; Bates, J.L.; Gerardy, K.; Linhares, D.C.L.; Piñeyro, P.E.; Schwartz, K.J.; Yoon, K.J.; Rademacher, C.J.; Holtkamp, D.; et al. Neonatal Mortality, Vesicular Lesions and Lameness Associated with Senecavirus A in a U.S. Sow Farm. Transbound. Emerg. Dis. 2016, 63, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Saeng-Chuto, K.; Rodtian, P.; Temeeyasen, G.; Wegner, M.; Nilubol, D. The first detection of Senecavirus A in pigs in Thailand, 2016. Transbound. Emerg. Dis. 2017, 65, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J.; Barcellos, D.; Alfieri, A.; Burrough, E.; Marthaler, D. Senecavirus A: An Emerging Pathogen Causing Vesicular Disease and Mortality in Pigs? Vet. Pathol. 2017, 54, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.E.S.; Michelazzo, M.M.Z.; Fernandes, T.; De Oliveira, A.G.; Leme, R.D.A.; Alfieri, A.F.; Alfieri, A.A.; Headley, S. Histopathological, immunohistochemical, and ultrastructural evidence of spontaneous Senecavirus A-induced lesions at the choroid plexus of newborn piglets. Sci. Rep. 2017, 7, 16555. [Google Scholar] [CrossRef]
- Dvorak, C.M.T.; Akkutay-Yoldar, Z.; Stone, S.R.; Tousignant, S.J.P.; Vannucci, F.; Murtaugh, M.P. An indirect enzyme-linked immunosorbent assay for the identification of antibodies to Senecavirus A in swine. BMC Veter Res. 2017, 13, 50. [Google Scholar] [CrossRef]
- Goolia, M.; Vannucci, F.; Patnayak, D.; Nfon, C.; Yang, M.; Babiuk, S. Validation of a competitive ELISA and a virus neutralization test for the detection and confirmation of antibodies to Senecavirus A in swine sera. J. Veter Diagn. Investig. 2017, 29, 250–253. [Google Scholar] [CrossRef]
- Fowler, V.L.; Ransburgh, R.H.; Poulsen, E.G.; Wadsworth, J.; King, D.P.; Mioulet, V.; Knowles, N.J.; Williamson, S.; Liu, X.; Anderson, G.A.; et al. Development of a novel real-time RT-PCR assay to detect Seneca Valley virus-1 associated with emerging cases of vesicular disease in pigs. J. Virol. Methods 2017, 239, 34–37. [Google Scholar] [CrossRef]
- Feronato, C.; Leme, R.D.A.; Diniz, J.A.; Agnol, A.M.D.; Alfieri, A.F.; Alfieri, A.F. Development and evaluation of a nested-PCR assay for Senecavirus A diagnosis. Trop. Anim. Health Prod. 2017, 50, 337–344. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Lin, X.; Chen, Z.; Wu, S. Development of a novel reverse transcription droplet digital PCR assay for the sensitive detection of Senecavirus A. Transbound. Emerg. Dis. 2019, 66, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jennison, A.; Whiley, D.; McMahon, J.; Hewitson, G.; Graham, R.; De Jong, A.; Warrilow, D.; Jennsion, A. Illumina sequencing of clinical samples for virus detection in a public health laboratory. Sci. Rep. 2019, 9, 5409. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Fan, W.; Qian, P.; Chen, H.; Li, X.-M. Isolation and full-genome sequencing of Seneca Valley virus in piglets from China, 2016. Virol. J. 2016, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, Q.; Bai, Y.; Chen, G.; Zhou, L.; Wu, Z.; Li, Y.; Zhou, W.; Yang, H.; Ma, J. Phylogenetic and genome analysis of seven senecavirus A isolates in China. Transbound. Emerg. Dis. 2017, 64, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Watson, M. Successful test launch for nanopore sequencing. Nat. Methods 2015, 12, 303–304. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.A.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Rambo-Martin, B.L.; Keller, M.W.; Wilson, M.M.; Nolting, J.M.; Anderson, T.K.; Vincent, A.L.; Bagal, U.; Jang, Y.; Neuhaus, E.B.; Davis, C.T.; et al. Influenza A virus field surveillance at a swine-human interface. mSphere 2020, 5, e00822-19. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Joshi, L.R.; Mohr, K.A.; Clement, T.; Hain, K.S.; Myers, B.; Yaros, J.; Nelson, E.A.; Christopher-Hennings, J.; Gava, D.; Schaefer, R.; et al. Detection of the emerging picornavirus Senecavirus A in pigs, mice, and houseflies. J. Clin. Microbiol. 2016, 54, 1536–1545. [Google Scholar] [CrossRef]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Schalamun, M.; Kainer, D.; Wang, W.; Schwessinger, B. MinIONQC: Fast and simple quality control for MinION sequencing data. Bioinformatics 2019, 35, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Juul, S.; Izquierdo, F.; Hurst, A.; Dai, X.; Wright, A.; Kulesha, E.; Pettett, R.; Turner, D.J. What’s in my pot? Real-time species identification on the MinION™. bioRxiv 2015, 030742. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Vaser, R.; Sovic, I.; Nagarajan, N.; Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhao, X.; Bai, Y.; Sun, B.; Xie, Q.; Ma, J. The First Identification and Complete Genome of Senecavirus A Affecting Pig with Idiopathic Vesicular Disease in China. Transbound. Emerg. Dis. 2017, 64, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, C.M.T.; Lilla, M.P.; Baker, S.R.; Murtaugh, M.P. Multiple routes of porcine circovirus type 2 transmission to piglets in the presence of maternal immunity. Vet. Microbiol. 2013, 166, 365–374. [Google Scholar] [CrossRef]
- Larkin, M.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.; McWilliam, H.; Valentin, F.; Wallace, I.; Wilm, A.; López, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Tan, S.; Dvorak, C.M.T.; Murtaugh, M.P. Rapid, Unbiased PRRSV Strain Detection Using MinION Direct RNA Sequencing and Bioinformatics Tools. Viruses 2019, 11, 1132. [Google Scholar] [CrossRef]
- Keller, M.W.; Rambo-Martin, B.L.; Wilson, M.M.; Ridenour, C.A.; Shepard, S.S.; Stark, T.J.; Neuhaus, E.B.; Dugan, V.G.; Wentworth, D.E.; Barnes, J. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Sci. Rep. 2018, 8, 14408. [Google Scholar] [CrossRef]
- Witwer, C.; Rauscher, S.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Picornaviridae genomes. Nucleic Acids Res. 2001, 29, 5079–5089. [Google Scholar] [CrossRef]
- Caliendo, A.M.; Gilbert, D.N.; Ginocchio, C.C.; Hanson, K.E.; May, L.; Quinn, T.C.; Tenover, F.C.; Alland, D.; Blaschke, A.J.; Bonomo, R.A.; et al. Better tests, better care: Improved diagnostics for infectious diseases. Clin. Infect. Dis. 2013, 57 (Suppl. 3), S139–S170. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Hersh, A.L.; Beekmann, S.E.; Ince, D.; Polgreen, L.A.; Hanson, K.E. Unmet Diagnostic Needs in Infectious Disease. Diagn. Microbiol. Infect. Dis. 2015, 81, 57–59. [Google Scholar] [CrossRef][Green Version]
- Woolhouse, M.E.; Brierley, L. Epidemiological characteristics of human-infective RNA viruses. Sci. Data 2018, 5, 180017. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Hernandez, R.; Jácome, R.; López-Vidal, Y.; De León, S.P. Are RNA viruses candidate agents for the next global pandemic? A review. ILAR J. 2017, 58, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Wongsurawat, T.; Jenjaroenpun, P.; Taylor, M.K.; Lee, J.; Tolardo, A.L.; Parvathareddy, J.; Kandel, S.; Wadley, T.D.; Kaewnapan, B.; Athipanyasilp, N.; et al. Rapid Sequencing of Multiple RNA Viruses in Their Native Form. Front. Microbiol. 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed]
- Ovcharenko, A.; Rentmeister, A. Emerging approaches for detection of methylation sites in RNA. Open Biol. 2018, 8, 180121. [Google Scholar] [CrossRef]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic Nanopore sequencing of influenza virus direct from clinical respiratory samples. J. Clin. Microbiol. 2019, 58, e00963-19. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Roberts, H.E.; Bonsall, D.; De Cesare, M.; Mokaya, J.; Lumley, S.F.; Golubchik, T.; Piazza, P.; Martin, J.B.; De Lara, C.; et al. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV). Sci. Rep. 2019, 9, 7081. [Google Scholar] [CrossRef]
- Davies, P.R. One world, one health: The threat of emerging swine diseases. A North American perspective. Transbound. Emerg. Dis. 2012, 59, 18–26. [Google Scholar] [CrossRef]
- Faria, N.R.; Sabino, E.C.; Nunes, M.R.T.; Alcantara, L.C.J.; Loman, N.J.; Pybus, O.G. Mobile real-time surveillance of Zika virus in Brazil. Genome Med. 2016, 8, 97. [Google Scholar] [CrossRef]
- Yamagishi, J.; Runtuwene, L.R.; Hayashida, K.; Mongan, A.E.; Thi, L.A.N.; Thuy, L.N.; Nhat, C.N.; Limkittikul, K.; Sirivichayakul, C.; Sathirapongsasuti, N.; et al. Serotyping dengue virus with isothermal amplification and a portable sequencer. Sci. Rep. 2017, 7, 3510. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Fauci, A.S. Emerging infectious diseases: Threats to human health and global stability. PLoS Pathog. 2013, 9, e1003467. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Sequencing Statistics | Direct RNA Sequencing (DRS) | PCR-cDNA Sequencing (PCS) |
|---|---|---|
| Number of available pores | 403 ± 100 | 421 ± 18 |
| Total pass yield (Mb) | 68 ± 0 | 92 ± 24 |
| Senecavirus A (SVA) yield (Mb) | 4.5 ± 0.6 | 66.1 ± 16.2 |
| Number of SVA reads | 3559 ± 358 | 38,544 ± 0 |
| Mean SVA read length | 1267 ± 40 | 1721 ± 47 |
| SVA read error rate (%) | 15.14 ± 0.32 | 11.23 ± 0.23 |
| Groups | Sequencing Statistics | Direct RNA Sequencing (DRS) | PCR-cDNA Sequencing (PCS) |
|---|---|---|---|
| Group 1 | Yield (Mb) | 68 ± 0 | 70 ± 0 |
| Read quality > 7 | Average length (bp) | 621 ± 2 | 1506 ± 19 |
| Average quality | 8.5 ± 0.4 | 8.6 ± 0.1 | |
| Group 2 | Yield (Mb) | 7.2 ± 0.4 | 52.6 ± 0.8 |
| Read quality >7+ | Average length (bp) | 2314 ± 35 | 2977 ± 24 |
| length > 1314 bp | Average quality | 8.6 ± 0 | 8.9 ± 0 |
| Group 3 | Yield (Mb) | 4.5 ± 0.6 | 66.1 ± 0.8 |
| Read quality >7+ | Average length (bp) | 1267 ± 40 | 1726 ± 49 |
| mapped to SVA database | Average quality | 8.3 ± 0.1 | 8.6 ± 0.1 |
| Group 1 | Group 2 (Length Filter) | Group 3 (SVA Mapped) | ||||||
|---|---|---|---|---|---|---|---|---|
| Sequencing method | Yield (Mb) | Consensus length (bp) | Accuracy (%) | Consensus length (bp) | Accuracy (%) | Consensus length (bp) | Accuracy (%) | SVA reads ^ |
| DRS | 0.7 | 5098 ± 786 | 89.3 ± 2.4 | 4881 ± 1728 | 89.8 ± 1.8 | 6057 ± 1143 | 86.2 ± 3.5 | 55 ± 2 |
| 7 | 7155 ± 4 | 90.8 ± 1.5 | 5522 ± 1229 | 91.2 ± 3 | 7091 ± 45 | 90.8 ± 1.3 | 548 ± 20 | |
| 70 | 7163 ± 18 | 94.3 ± 0.2 | 7096 ± 30 | 94.4 ± 0.4 | 7110 ± 21 | 94.4 ± 0.5 | 3559 ± 358 | |
| PCS | 0.7 | 6738 ± 424 | 99 ± 0.1 | 6316 ± 541 | 97.4 ± 2.4 | 6592 ± 489 | 98 ± 1.0 | 410 ± 6 |
| 7 | 7267 ± 132 | 98.9 ± 0.4 | 7238 ± 19 | 99.0 ± 0.1 | 7079 ± 161 | 99.0 ± 0.0 | 4092 ± 64 | |
| 70 | 3053 ± 35 | 90.5 ± 11.5 | 6596 ± 1903 | 91.6 ± 5.4 | 6761 ± 995 | 87.9 ± 2.0 | 38544 ± 10439 | |
| Species Detection | Strain Level Identification | Consensus Generation | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ct value | Viral copies | Total reads | WIMP ‡ SVA reads | GenBank reference sequence * | DRS | PCS | Minimap SVA reads | DRS consensus | PCS consensus | ||||||||
| DRS | PCS | DRS | PCS | Best match | Identity (%) | Best match | Identity (%) | DRS | PCS | Length ^ (bp) | Accuracy (%) | Length ^ (bp) | Accuracy (%) | ||||
| Spike-in samples | 10 | 1.0 × 107 | 94,572 | 16,515 | 5722 | 2913 | MN164664 | MN164664 | 100.0 | MN164664 | 100.0 | 42,936 | 4658 | 7189 | 94.8 | 7395 | 99.2 |
| 13 | 1.2 × 106 | 40,555 | 5775 | 236 | 1182 | MN164664 | MN164664 | 100.0 | MN164664 | 100.0 | 1961 | 1611 | 7177 | 91.0 | 6873 | 99.0 | |
| 18 | 6.5 × 104 | 185,259 | 5071 | 17 | 296 | MN164664 | MN164664 | 100.0 | MN164664 | 100.0 | 43 | 420 | 2551 | 90.6 | 6770 | 99.4 | |
| 20 | 1.2 × 104 | 219,789 | 3889 | 3 | 3 | MN164664 | MN164664 | 100.0 | MN164664 | 100.0 | 10 | 3 | 1306 | 90.3 | 916 † | 80.8 | |
| 25 | 4.7 × 102 | 110,641 | 7880 | 1 | 0 | MN164664 | MN164664 | 100.0 | NA | NA | 1 | 0 | 456 † | 85.1 | NA | NA | |
| Clinical Samples | 13 | 1.1 × 106 | 45,478 | 78,075 | 299 | 436 | MN990489 | KX019804.1 | 97.1 | KX019804.1 | 97.1 | 377 | 630 | 7157 | 97.0 | 7013 | 99.6 |
| 16 | 1.3 × 105 | 148,513 | 5256 | 83 | 7 | MN990490 | KX019804.1 | 98.2 | KU058182.1 | 97.8 | 124 | 9 | 3421 | 96.4 | 880 † | 89.0 | |
| 18 | 5.0 × 104 | 47,411 | 174,551 | 19 | 24 | MN990491 | KY618836.1 | 97.4 | KY618836.1 | 97.4 | 20 | 27 | 6534 | 97.1 | 2285 † | 95.3 | |
| 20 | 1.2 × 104 | 54,964 | 5943 | 3 | 0 | MN990492 | KU051394.1 | 97.9 | NA | 4 | 0 | 1957 | 82.4 | NA | NA | ||
| 21 | 7.6 × 103 | 39,465 | 242,239 | 3 | 4 | MN990493 | KY618835.1 | 97.7 | MK256736.1 | 97.4 | 3 | 5 | 1158 | 94.3 | 988 † | 87.0 | |
| 22 | 2.3 × 103 | 53,359 | 96,661 | 1 | 4 | MN990494 | KY618836.1 | 97.4 | KT827250.1 | 97.7 | 1 | 7 | 511 † | 88.8 | 7745 † | 85.0 | |
| 23 | 2.2 × 103 | 58,929 | 633,632 | 4 | 1 | MN990495 MN997126 | KX019804.1 | 98.2 | MH634514.1 | 98.2 | 4 | 1 | 3206 | 92.1 | 1171 † | 94.5 | |
| 24 | 9.2 × 102 | 41,645 | 135,552 | 1 | 0 | MN990496 | MH490944.1 | 97.0 | NA | NA | 1 | 0 | 300 † | 80.0 | NA | NA | |
| Direct RNA Sequencing (DRS) | PCR-cDNA Sequencing (PCS) | |
|---|---|---|
| Laboratory time (sample prep) | 3 h | 5 h |
| Sequencing time | 6 h | 6 h |
| Amount of RNA recommended for input | 500 ng * | 2 ng |
| Analytical sensitivity (viral copies) | 102 to 103 | 103 to 104 |
| Recommended consensus generation program | Racon | Canu |
| Raw read accuracy | 85% | 89% |
| Consensus accuracy | 94% | 99% |
| Consensus genome coverage ^ | 100% | 100% |
| Read distribution | Coverage biases | Even coverage |
| Key attributes | Rapid, sensitive, potential RNA structure detection | Accurate |
| Key concerns | High input RNA amount *, higher error rate | Need for amplification, longer time to results |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, S.; Dvorak, C.M.T.; Murtaugh, M.P. Characterization of Emerging Swine Viral Diseases through Oxford Nanopore Sequencing Using Senecavirus A as a Model. Viruses 2020, 12, 1136. https://doi.org/10.3390/v12101136
Tan S, Dvorak CMT, Murtaugh MP. Characterization of Emerging Swine Viral Diseases through Oxford Nanopore Sequencing Using Senecavirus A as a Model. Viruses. 2020; 12(10):1136. https://doi.org/10.3390/v12101136
Chicago/Turabian StyleTan, Shaoyuan, Cheryl M. T. Dvorak, and Michael P. Murtaugh. 2020. "Characterization of Emerging Swine Viral Diseases through Oxford Nanopore Sequencing Using Senecavirus A as a Model" Viruses 12, no. 10: 1136. https://doi.org/10.3390/v12101136
APA StyleTan, S., Dvorak, C. M. T., & Murtaugh, M. P. (2020). Characterization of Emerging Swine Viral Diseases through Oxford Nanopore Sequencing Using Senecavirus A as a Model. Viruses, 12(10), 1136. https://doi.org/10.3390/v12101136