A Functional K+ Channel from Tetraselmis Virus 1, a Member of the Mimiviridae

 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
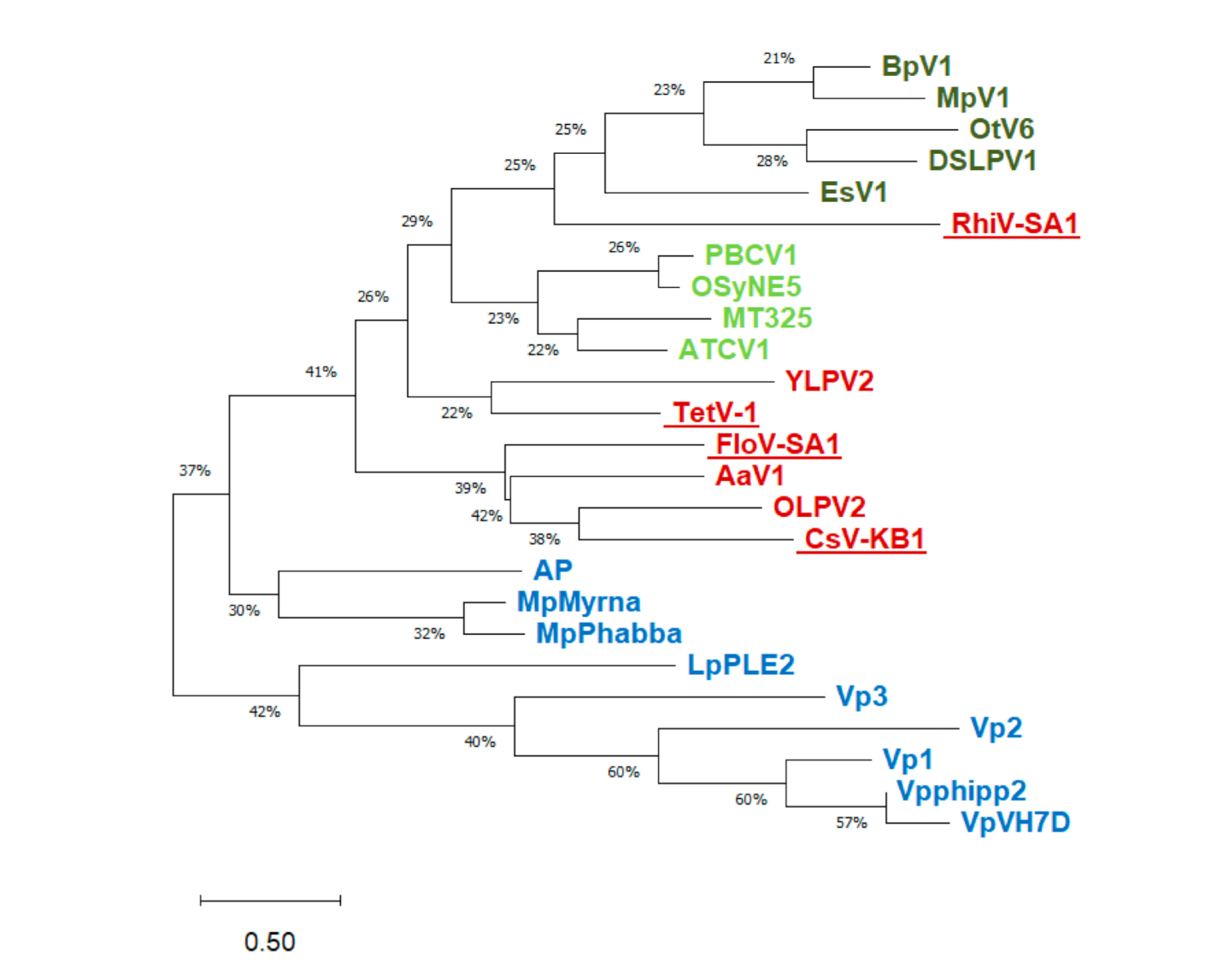
2.1. Sequence Sources
2.2. Sequence Analysis
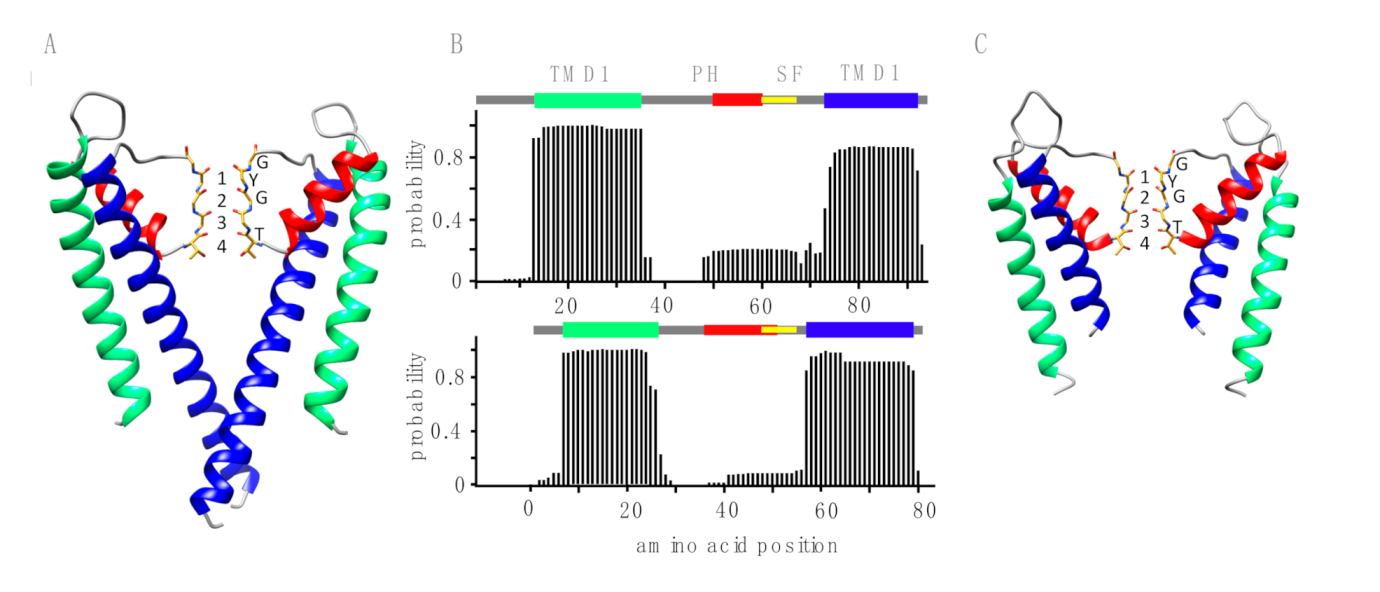
2.3. 3D Modeling
2.4. Planar Lipid Bilayer Experiments
2.5. Channel Protein Synthesis
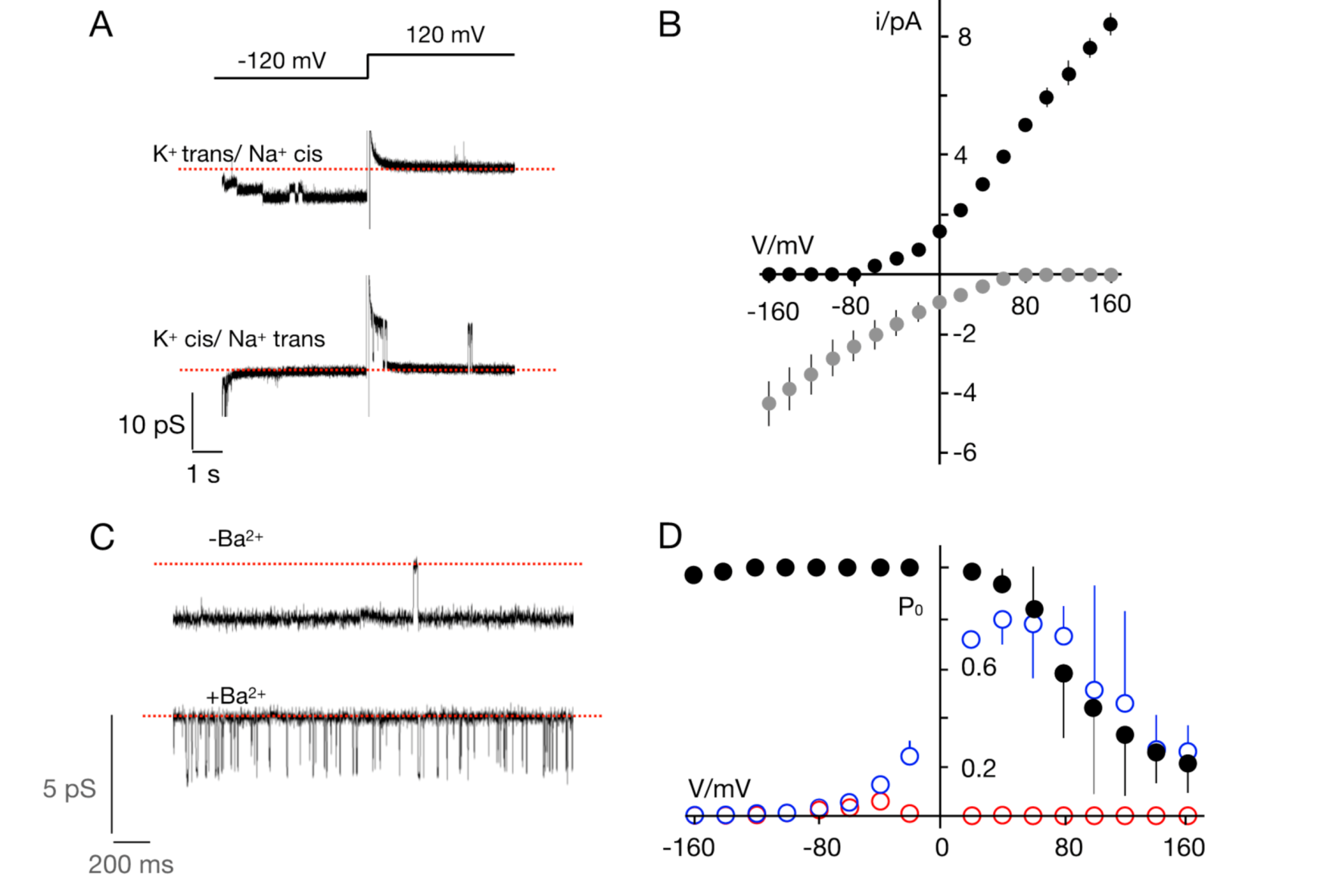
2.6. Ion Channel Recordings, Data Analysis, and Statistics
3. Results and Discussion
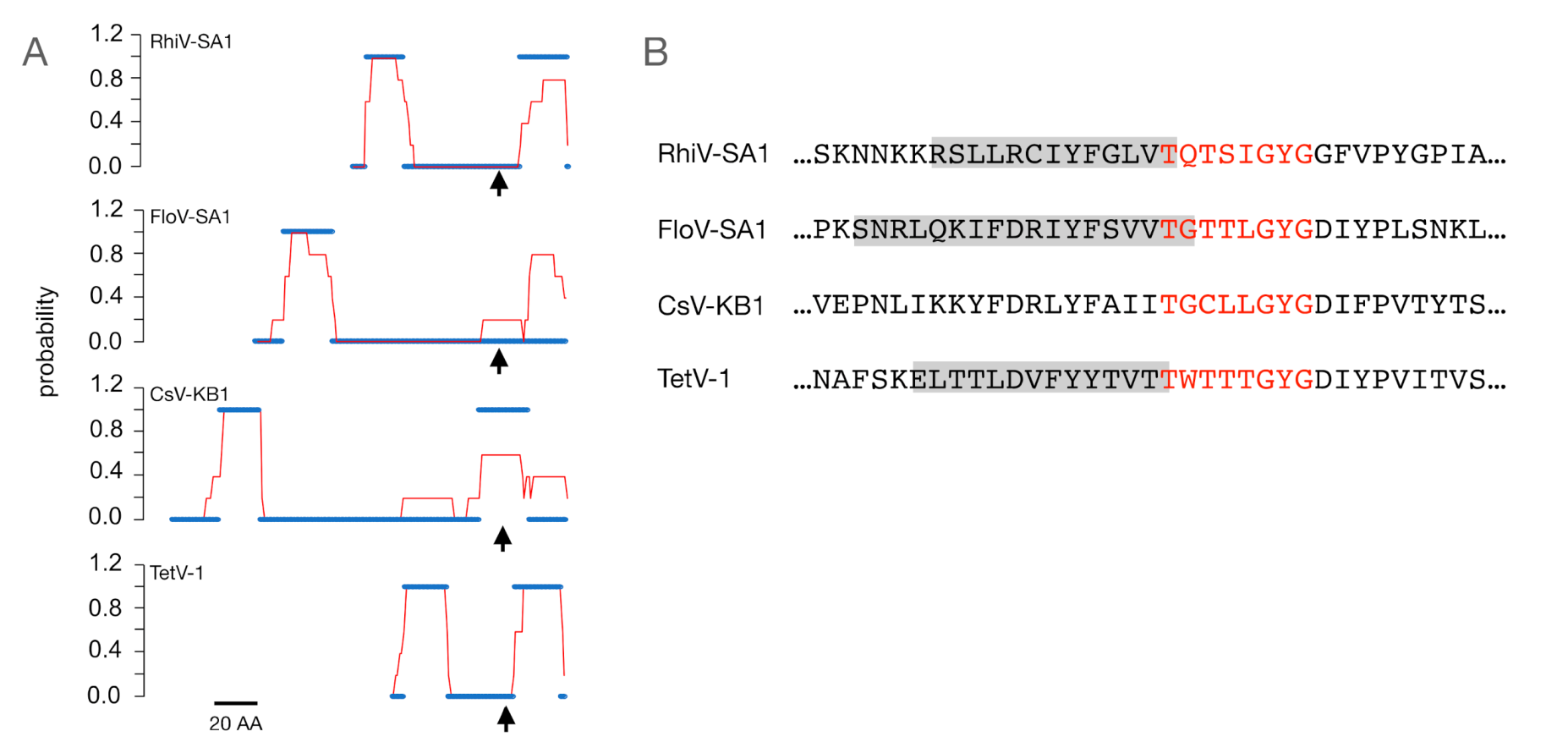
3.1. Putative K+ Channels
3.2. The TetV-1 Protein Has K+ Channel Function
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Fischer, W.B.; Sansom, M.S. Viral ion channels: Structure and function. Biochim. Biophys. Acta 2002, 1561, 27–45. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Carrasco, L. Viroporins. FEBS Lett. 2003, 552, 28–34. [Google Scholar] [CrossRef]
- Wang, K.; Xie, S.Q.; Sun, B. Viral proteins function as ion channels. Biochim. Biophys. Acta 2011, 1808, 510–515. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Murry, C.; Agarkova, I.V.; Ghosh, J.; Fitzgerald, F.; Carlson, R.; Hertel, B.; Kukovetz, K.; Rauh, O.; Thiel, G.; Van Etten, J.L. Genetic diversity of potassium ion channel proteins encoded by chloroviruses that infect Chlorella heliozoae. Viruses 2020, 12, 678. [Google Scholar] [CrossRef]
- Plugge, B.; Gazzarini, S.; Cerana, R.; Van Etten, J.; Nelson, M.; DiFrancesco, D.; Moroni, A.; Thiel, G. A potassium ion channel protein encoded by chlorella virus PBCV-1. Science 2000, 287, 1641–1644. [Google Scholar] [CrossRef]
- Greiner, T.; Moroni, A.; Van Etten, J.L.; Thiel, G. Genes for membrane transport proteins: Not so rare in viruses. Viruses 2018, 10, 456. [Google Scholar] [CrossRef]
- Tayefeh, S.; Kloss, T.; Kreim, M.; Gebhardt, M.; Baumeister, D.; Hertel, B.; Richter, C.; Schwalbe, H.; Moroni, A.; Thiel, G.; et al. Model development for the viral Kcv potassium channel. Biophys. J. 2009, 96, 485–498. [Google Scholar] [CrossRef]
- Thiel, G.; Baumeister, D.; Schroeder, I.; Kast, S.M.; Van Etten, J.L.; Moroni, A. Minimal art: Or why small viral K+ channels are good tools for understanding basic structure and function relations. Biochim. Biophys. Acta 2011, 1808, 580–588. [Google Scholar] [CrossRef]
- Thiel, G.; Moroni, A.; Blanc, G.; Van Etten, J.L. Potassium ion channels: Could they have evolved from viruses? Plant Physiol. 2013, 162, 1215–1224. [Google Scholar] [CrossRef]
- Rauh, O.; Urban, M.; Henkes, L.M.; Winterstein, T.; Greiner, T.; Van Etten, J.L.; Moroni, A.; Kast, S.M.; Thiel, G.; Schroeder, I. Sequence specific distortions of a transmembrane helix generate a gate in a K+ channel with a long lived closed state. J. Am. Chem. Soc. 2017, 139, 7494–7503. [Google Scholar] [CrossRef]
- Rauh, O.; Hansen, U.P.; Scheub, D.D.; Thiel, G.; Schroeder, I. Site-specific ion occupation in the selectivity filter causes voltage-dependent gating in a viral K+ channel. Sci. Rep. 2018, 8, 10406. [Google Scholar] [CrossRef]
- Kang, M.; Moroni, A.; Gazzarrini, S.; DiFrancesco, D.; Thiel, G.; Severino, M.; Van Etten, J.L.; Severino, M.; Van Etten, J.L. Small potassium ion channel protein encoded by chlorella viruses. Proc. Natl. Acad. Sci. USA 2004, 101, 5318–5324. [Google Scholar] [CrossRef]
- Gazzarrini, S.; Kang, M.; Epimashko, S.; Van Etten, J.L.; Dainty, J.; Thiel, G.; Moroni, A. Chlorella virus MT325 encodes water and potassium channels that interact synergistically. Proc. Natl. Acad. Sci. USA 2006, 103, 5355–5360. [Google Scholar] [CrossRef]
- Balss, J.; Mehmel, M.; Baumeister, D.; Hertel, B.; Delaroque, N.; Chatelain, F.C.; Minor, D.J.; Van Etten, J.L.; Moroni, A.; Thiel, G. Transmembrane domain length of viral K+ channels is a signal for mitochondria targeting. Proc. Natl. Acad. Sci. USA 2008, 105, 12313–12318. [Google Scholar] [CrossRef]
- Siotto, F.; Martin, C.; Rauh, O.; Van Etten, J.L.; Schroeder, I.; Moroni, A.; Thiel, G. Viruses infecting marine picoplancton encode functional potassium ion channels. Virology 2014, 467, 103–111. [Google Scholar] [CrossRef]
- Hamacher, K.; Greiner, T.; Van Etten, J.L.; Gebhardt, M.; Cosentino, C.; Moroni, A.; Thiel, G. Phycodnavirus potassium ion channel proteins question the virus molecular piracy hypothesis. PLoS ONE 2012, 7, e38826. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; LeCleir, G.R.; Brown, C.M.; Gobler, C.J.; Bidle, K.D.; Wilson, W.H.; Wilhelm, S.W. Genome of brown tide virus (AaV), the little giant of the Megaviridae, elucidates NCLDV genome expansion and host-virus coevolution. Virology 2014, 466–467, 60–70. [Google Scholar] [CrossRef]
- Schvarcz, C.R.; Steward, G.F. A giant virus infecting green algae encodes key fermentation genes. Virology 2018, 518, 423–433. [Google Scholar] [CrossRef]
- Yau, S.; Lauro, F.M.; DeMaere, M.Z.; Brown, M.V.; Thomas, T.; Raftery, M.J.; Andrews-Pfannkoch, C.; Lewis, M.; Hoffman, J.M.; Gibson, J.A.; et al. Virophage control of antarctic algal host-virus dynamics. Proc. Natl. Acad. Sci USA 2011, 108, 6163–6168. [Google Scholar] [CrossRef]
- Winterstein, L.M.; Kukovetz, K.; Rauh, O.; Turman, D.L.M.; Braun, C.; Moroni, A.; Schroeder, I.; Thiel, G.; Turman, D.L.M.; Braun, C.; et al. Reconstitution and functional characterization of ion channels from nanodiscs in lipid bilayers. J. Gen. Physiol. 2018, 150, 637–646. [Google Scholar] [CrossRef]
- Schvarcz, C.R. Cultivation and Characterization of Viruses Infecting Eukaryotic Phytoplankton from the Tropical North Pacific Ocean. Doctoral Dissertation, University of Hawai’i at Mānoa, Honolulu, HI, USA, 2019. Available online: http://hdl.handle.net/10125/62656 (accessed on 14 August 2020).
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translationinitiation site identification. BMC Bioinform. 2020, 11, 119. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Weissgraeber, S.; Saponaro, A.; Thiel, G.; Hamacher, K. A reduced mechanical model for cAMP modulated gating in HCN channels. Sci. Rep. 2017, 7, 40168. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Shealy, T.R.; Murphy, A.D.; Ramarathnam, R.; Jakobsson, E.; Subramanian, S. Sequence-function analysis of the K-selective family of ion channels using a comprehensive alignment and the KcsA channel structure. Biophys. J. 2003, 84, 2929–2942. [Google Scholar] [CrossRef]
- Doyle, D.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatic 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Watanabe, Y.; Martini, J.E.; Ohmoto, H. Geochemical evidence for terrestrial ecosystems 2.6 billion years ago. Nature 2000, 408, 574–578. [Google Scholar] [CrossRef]
- Lewin, G.R.; Carlos, C.; Chevrette, M.G.; Horn, H.A.; McDonald, B.R.; Stankey, R.J.; Fox, B.G.; Currie, C.R. Evolution and ecology of Actinobacteria and their bioenergy applications. Annu. Rev. Microbiol. 2016, 70, 235–254. [Google Scholar] [CrossRef]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient virus world and evolution of cells. Biol. Direct. 2006, 29. [Google Scholar] [CrossRef]
- Guglielmini, J.; Woo, A.C.; Krupovic, M.; Forterre, P.; Gaia, M. Diversification of giant and large eukaryotic dsDNA viruses predated the origin of modern eukaryotes. Proc. Natl. Acad. Sci. USA 2019, 116, 19585–19592. [Google Scholar] [CrossRef]
- Braun, C.J.; Lachnit, C.; Becker, P.; Henkes, L.M.; Arrigoni, C.; Kast, S.M.; Moroni, A.; Thiel, G.; Schroeder, I. Viral potassium channels as a robust model system for studies of membrane-protein interaction. Biochim. Biophys. Acta 2014, 1838, 1096–1103. [Google Scholar] [CrossRef]
- Valiyaveetil, F.I.; Zhou, Y.; MacKinnon, R. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry 2002, 41, 10771–10777. [Google Scholar] [CrossRef]
- Rondelli, V.; Del Favero, E.; Brocca, P.; Fragneto, G.; Trapp, M.; Mauri, L.; Ciampa, M.G.; Romani, G.; Braun, C.J.; Schroeder, I.; et al. Vectional K+ Channel insertion in a single phospholipid bilayer: Neutron reflectometry and electrophysiology in the joint exploration of a model membrane functional platform. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 1742–1750. [Google Scholar] [CrossRef]
- Hille, B. Ion Channels of Excitable Membranes, 3th ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2001. [Google Scholar]
- Piasta, K.N.; Theobald, D.L.; Millerm, C. Potassium-selective block of barium permeation through single KcsA channels. J. Gen. Physiol. 2011, 138, 421–436. [Google Scholar] [CrossRef]
- Sohma, Y.; Harris, A.; Wardle, C.J.C.; Argent, B.R.E.; Gray, M.A. Two barium binding sites on a maxi K+ channel from human vas deferens epithelial cells. Biophys. J. 1996, 70, 1316–1325. [Google Scholar] [CrossRef]
- Colson, P.; La Scola, B.; Levasseur, A.; Caetano-Anolles, G.; Raoult, D. Mimivirus: Leading the way in the discovery of giant viruses of amoebae. Nat. Rev. Microbiol. 2017, 15, 243–253. [Google Scholar] [CrossRef]
- Strandberg, E.; Killian, J.A. Snorkeling of lysine side chains in transmembrane helices: How easy can it get? FEBS Lett. 2003, 544, 69–73. [Google Scholar] [CrossRef]
- Zakharian, E. Recording of ion channel activity in planar lipid bilayer experiments. Methods Mol. Biol. 2013, 998, 109–118. [Google Scholar]
- Hartel, A.J.W.; Shekar, S.; Ong, P.; Schroeder, I.; Thiel, G.; Shepard, K.L. High bandwidth approaches in nanopore and ion channel recordings—A tutorial review. Anal. Chim. Acta 2019, 1061, 13–27. [Google Scholar] [CrossRef]
- Sandaa, R.-A.; Dahle, H.; Brussaard, C.P.D.; Ogata, H.; Blanc-Mathieu, R. Algal viruses belonging to a subgroup within the Mimiviridae family. Ref. Modul. Life Sci. 2020. [Google Scholar] [CrossRef]
- Thiel, G.; Moroni, A.; Dunigan, D.; Van Etten, J.L. Initial Events Associated with Virus PBCV-1 Infection of Chlorella NC64A. Prog. Bot. 2010, 71, 169–183. [Google Scholar]
- Lee, S.W.; Lee, H.H.; Thiel, G.; Van Etten, J.L.; Saraf, R.F. Noninvasive measurement of a single viral infection in a cell. ACS Nano 2016, 10, 5123–5130. [Google Scholar] [CrossRef]
- Kirst, G.O.; Bisson, M. Regulation of turgor pressure in marine algae: Ions and low molecular-weight organic compounds. Funct. Plant Biol. 2029, 6, 539–556. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, T.K. Viral calciomics: Interplay between Ca2+ and virus. Cell Calcium 2009, 46, 1–7. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Putative Channel Protein (NCBI Accession #) | Most Similar Non-Virus Hit, Organism Max Score, Total Score, Query Cover, E-Value, aa Identity, Accession # | Most Similar Virus Hit, Virus Max Score, Total Score, Query Cover, E-Value, aa Identity, Accession # |
|---|---|---|
| RhiV-SA1 (MT926120) | Oleiphilus messinensis (Proteobacteria) 49.7, 49.7, 72%, 2 × 10−4, 36%, WP_087462234.1 | Acanthocystis turfacea chlorella virus mid_1.1 ATCV-1 (Chlorovirus) 43.9, 43.9, 46%, 4 × 10−5, 34%, QLC35876.1 |
| FloV-SA1 (MT926122) | Actinomycetales bacterium (Actinobacteria) 77.8, 77.8, 88%, 3 × 10−15, 38%, PQM59864.1 | Aureococcus anophagefferens virus (Aav) 58.2, 58.2, 90%, 1 × 10−9, 33%, YP_009052228.1 |
| CsV-KB1 (MT926121) | Actinomycetales bacterium (Actinobacteria) 93.2, 93.2, 86%, 8 × 10−21, 36%, PQM59864.1 | Organic Lake phycodnavirus 2 (OLPV2) 82.8, 82.8, 76%, 4 × 10−19, 39%, ADX06223.1 |
| TetV-1 (AUF82121.1) | Streptomyces laurentii (Actinobacteria) 55.5, 55.5, 67%, 4 × 10−7, 42%, BAU83004.1 | Ostreococcus tauri virus RT-201 40.8, 40.8, 61%, 4 × 10−4, 41%, AFC34969.1 |
| Source of Putative Channel Protein | nc/na in DPhPC | nc/na in DPhPG |
|---|---|---|
| RhiV-SA1 | 0/3 | not tested |
| FloV-SA1 | 0/3 | 0/1 |
| CsV-KB1 | 0/3 | not tested |
| TetV-1 | 39/39 | not tested |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kukovetz, K.; Hertel, B.; Schvarcz, C.R.; Saponaro, A.; Manthey, M.; Burk, U.; Greiner, T.; Steward, G.F.; Van Etten, J.L.; Moroni, A.; et al. A Functional K+ Channel from Tetraselmis Virus 1, a Member of the Mimiviridae. Viruses 2020, 12, 1107. https://doi.org/10.3390/v12101107
Kukovetz K, Hertel B, Schvarcz CR, Saponaro A, Manthey M, Burk U, Greiner T, Steward GF, Van Etten JL, Moroni A, et al. A Functional K+ Channel from Tetraselmis Virus 1, a Member of the Mimiviridae. Viruses. 2020; 12(10):1107. https://doi.org/10.3390/v12101107
Chicago/Turabian StyleKukovetz, Kerri, Brigitte Hertel, Christopher R. Schvarcz, Andrea Saponaro, Mirja Manthey, Ulrike Burk, Timo Greiner, Grieg F. Steward, James L. Van Etten, Anna Moroni, and et al. 2020. "A Functional K+ Channel from Tetraselmis Virus 1, a Member of the Mimiviridae" Viruses 12, no. 10: 1107. https://doi.org/10.3390/v12101107
APA StyleKukovetz, K., Hertel, B., Schvarcz, C. R., Saponaro, A., Manthey, M., Burk, U., Greiner, T., Steward, G. F., Van Etten, J. L., Moroni, A., Thiel, G., & Rauh, O. (2020). A Functional K+ Channel from Tetraselmis Virus 1, a Member of the Mimiviridae. Viruses, 12(10), 1107. https://doi.org/10.3390/v12101107