A New Approach to 3D Modeling of Inhomogeneous Populations of Viral Regulatory RNA


Abstract
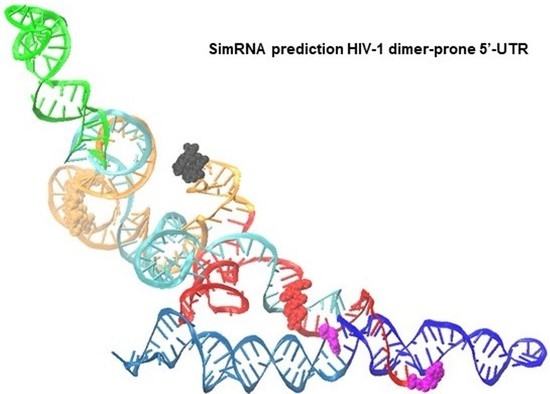
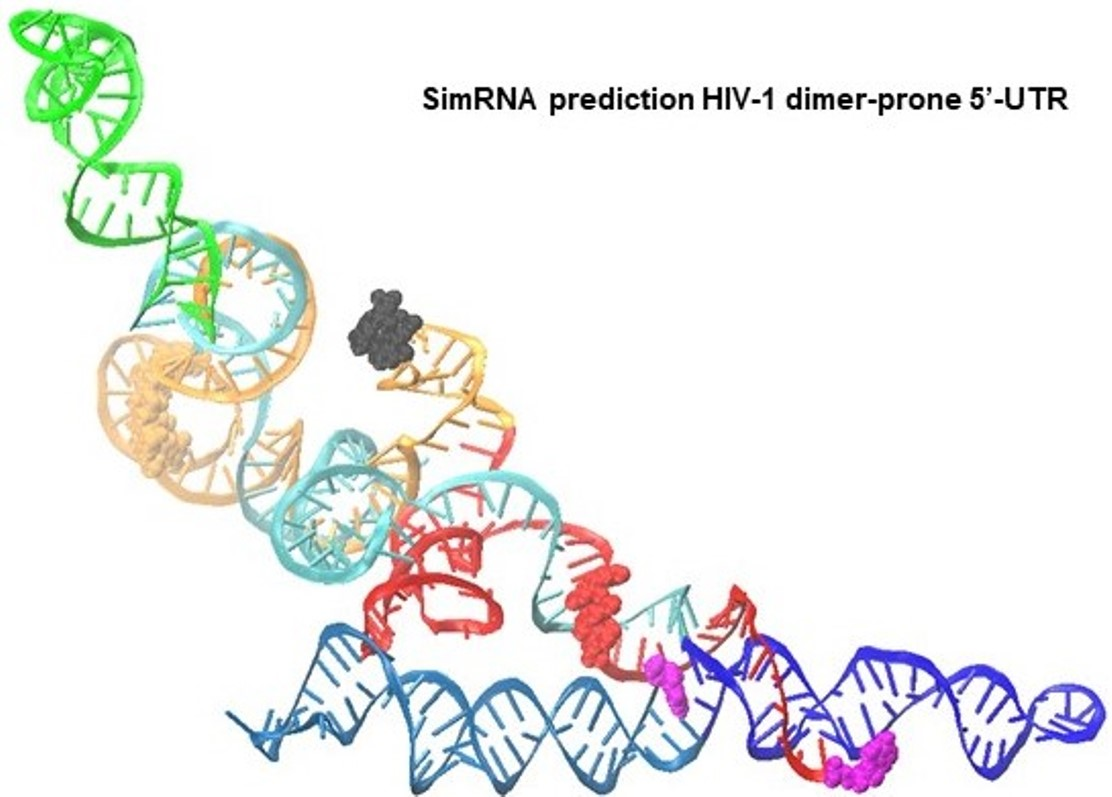
1. Introduction
2. Materials and Methods
3. Results
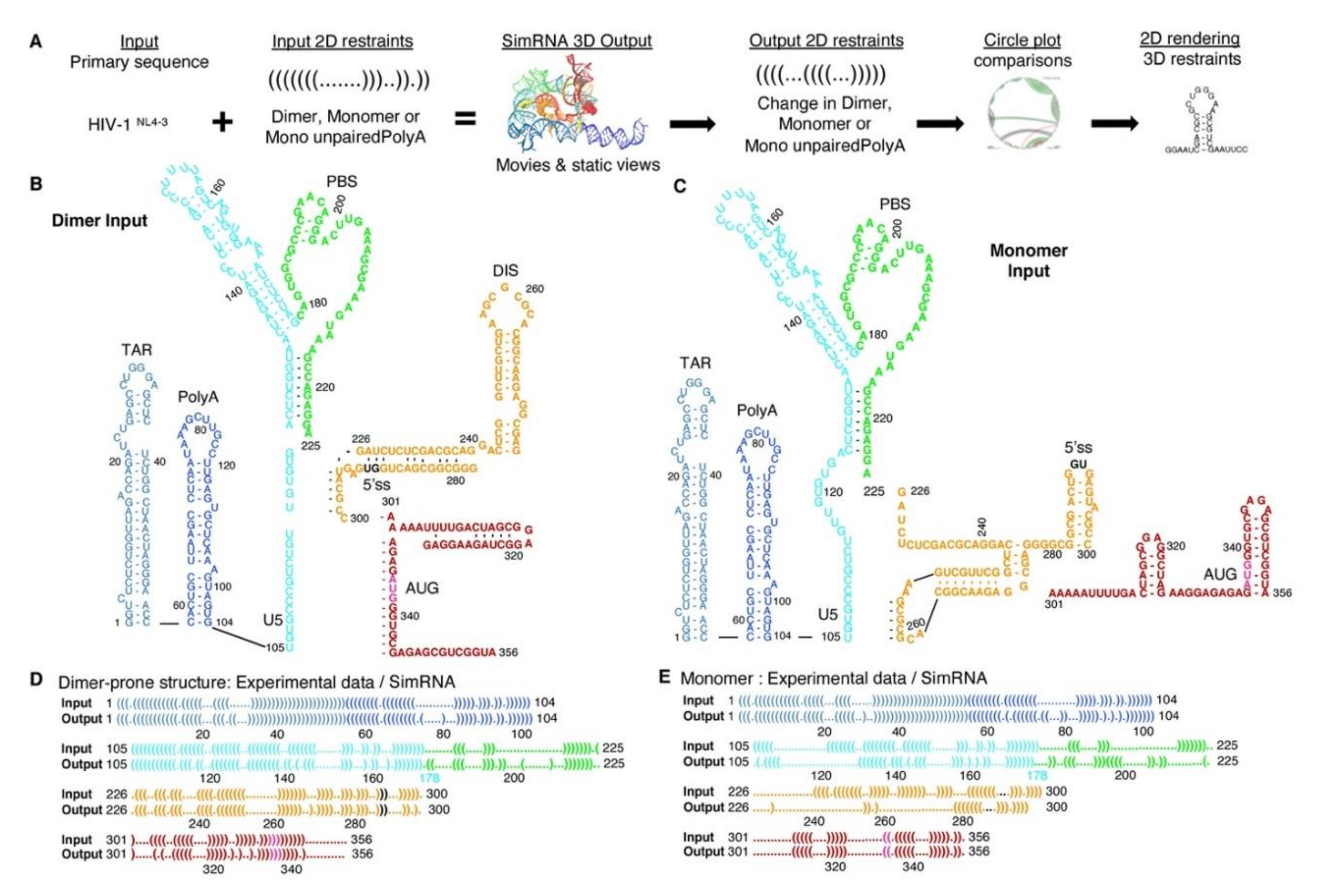
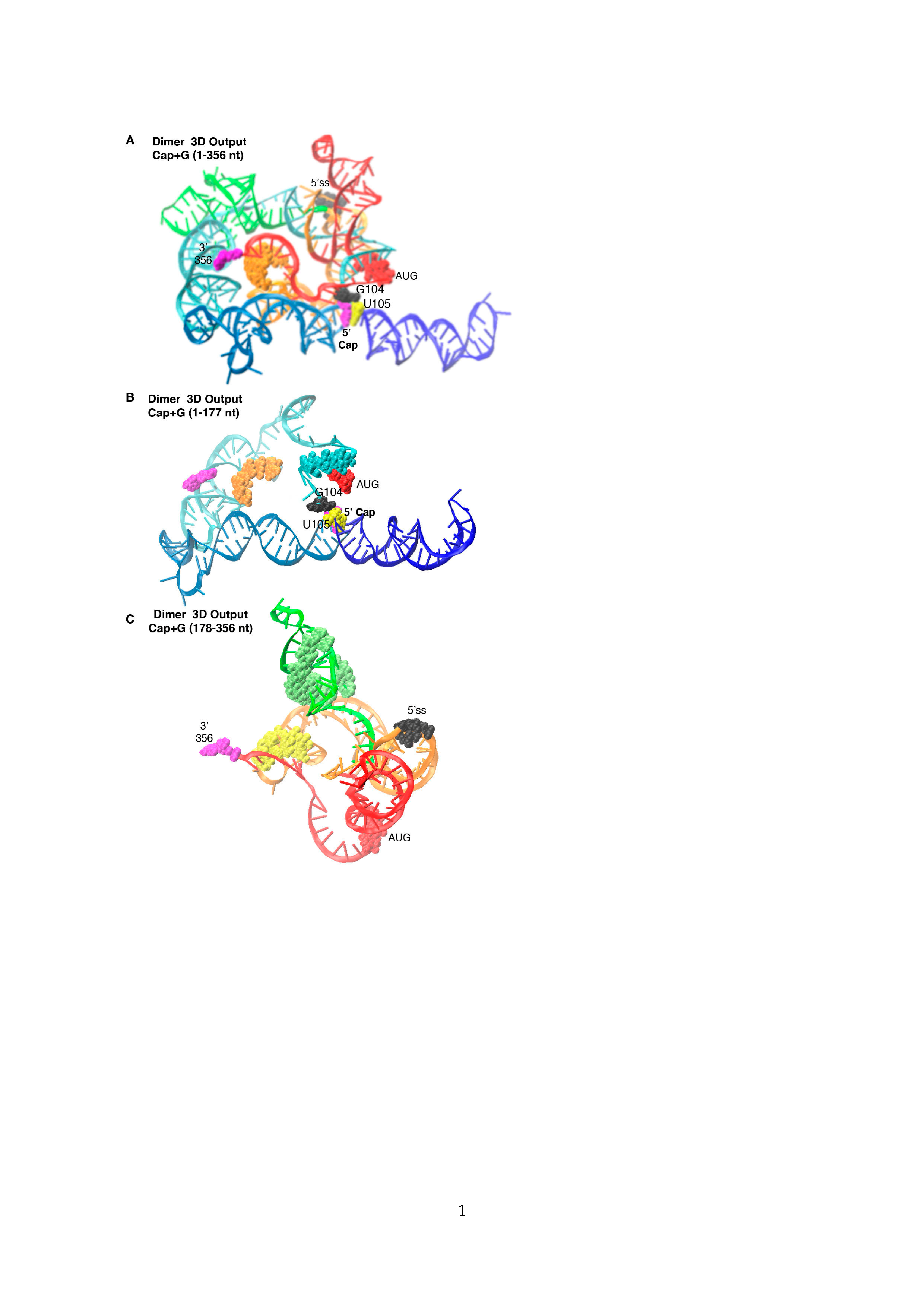
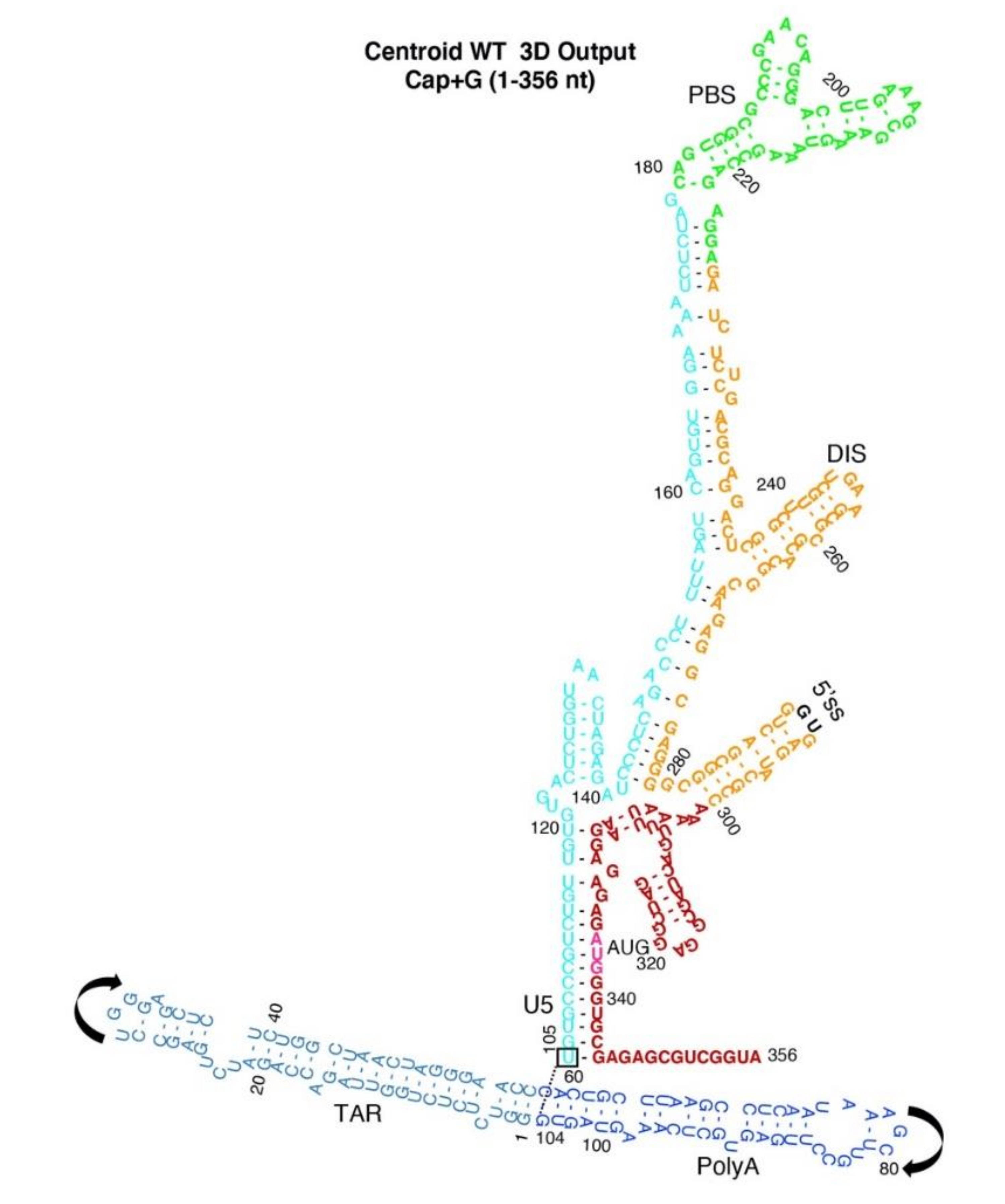
3.1. SimRNA Evaluated 3D Properties of HIVNL4-3 5′-UTR Beginning with 5′-Capped-Guanosine
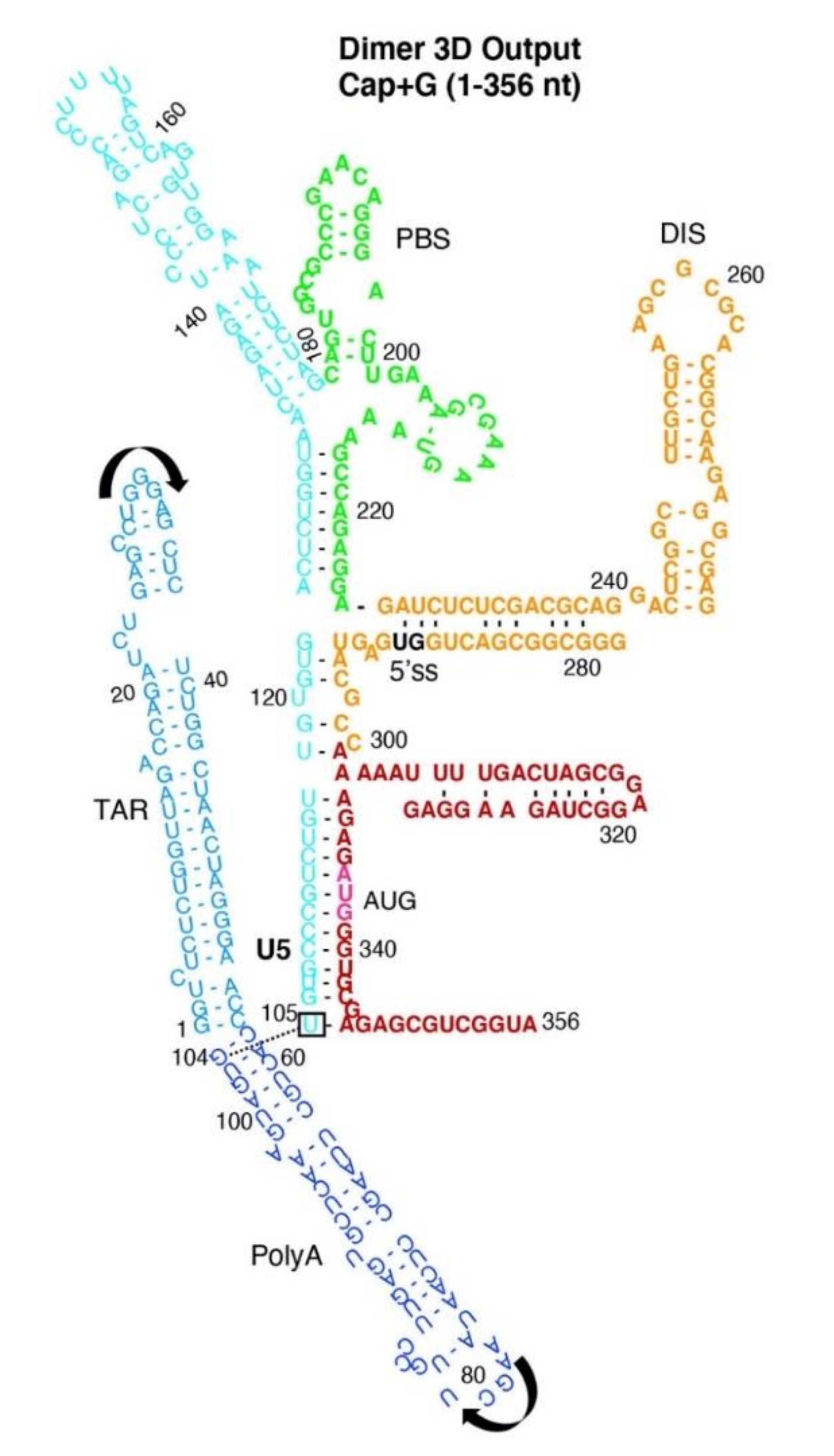
3.2. Dimer Output 3D Model Predicted TAR-PolyA-U5 Converge near the 5′-Cap
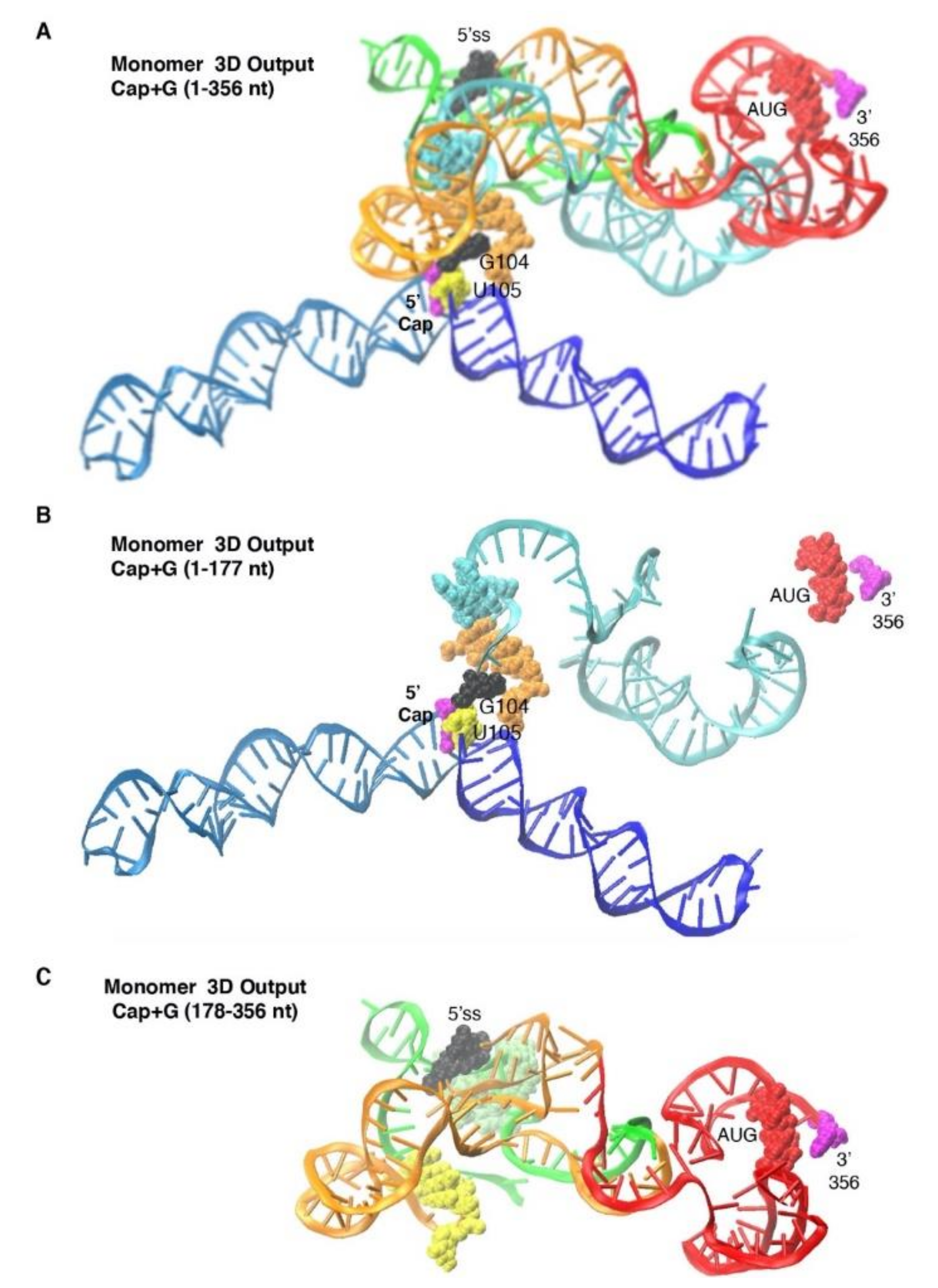
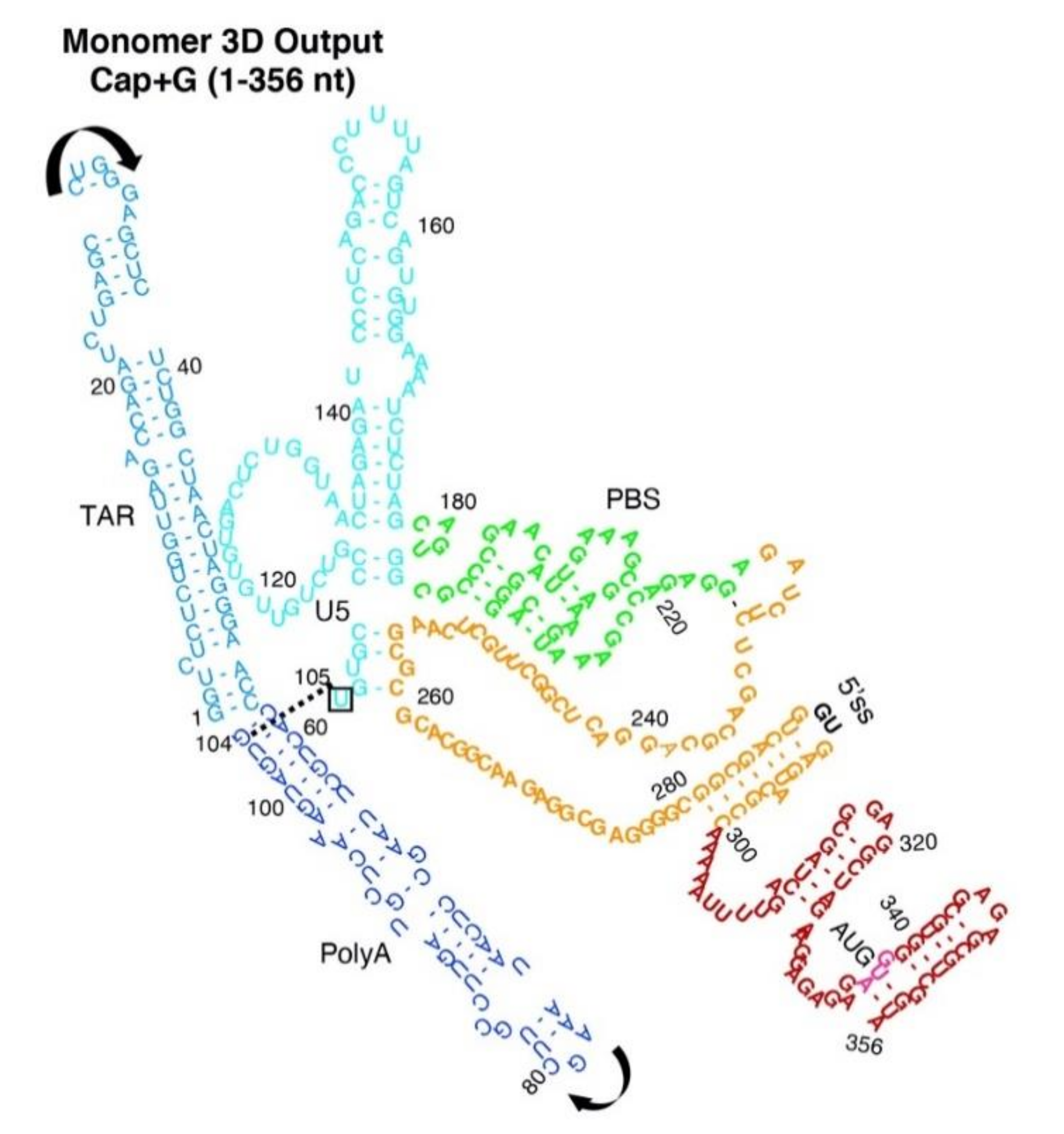
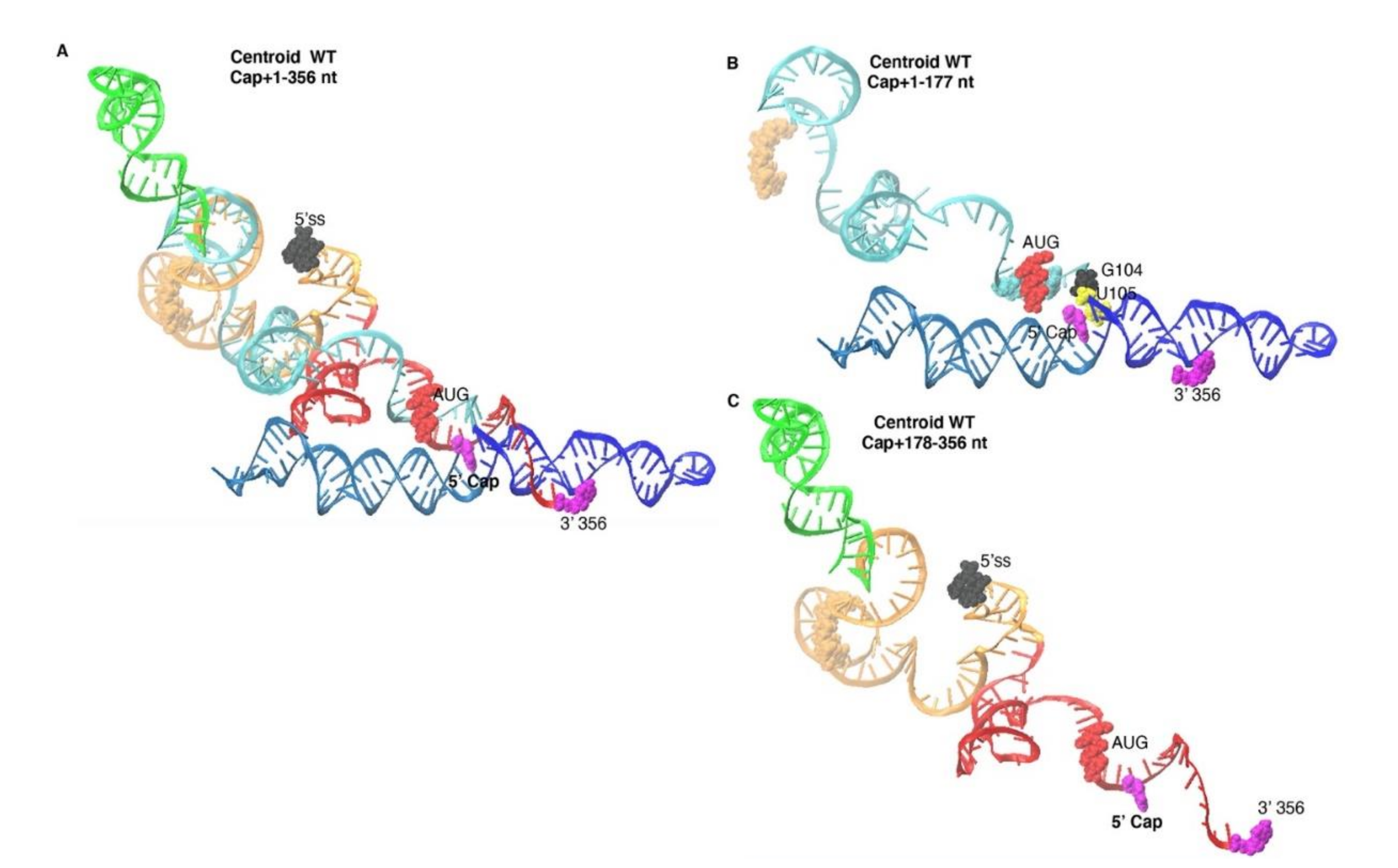
3.3. Monomer Output 3D Model Altered Accessibility of 5′-Cap and 5′-ss and Reoriented U5 Stem
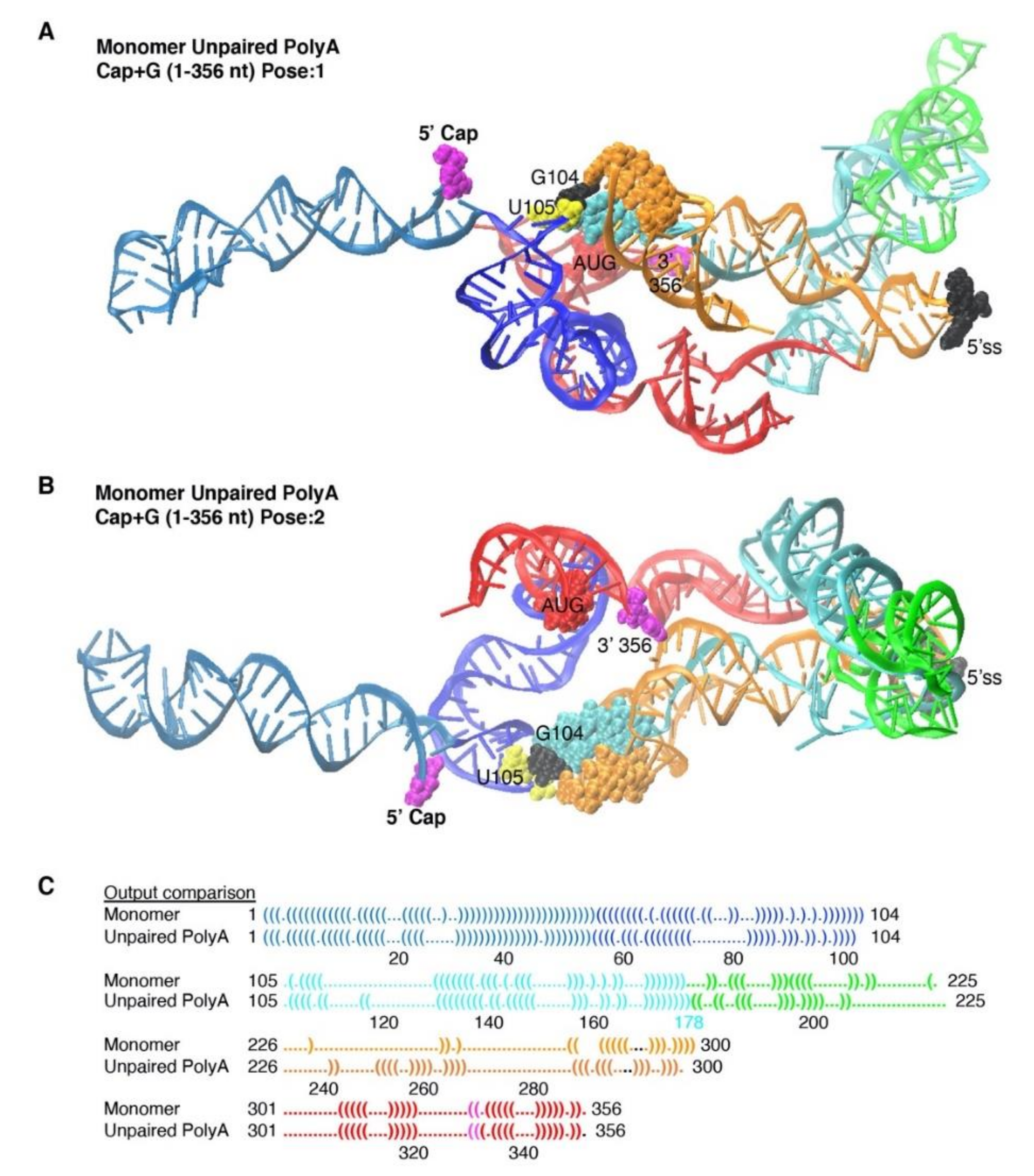
3.4. The Unpairing of PolyA Nts Reduced Local Energy Minima in the Thermodynamic Equilibrium to Monomer Tertiary Structure
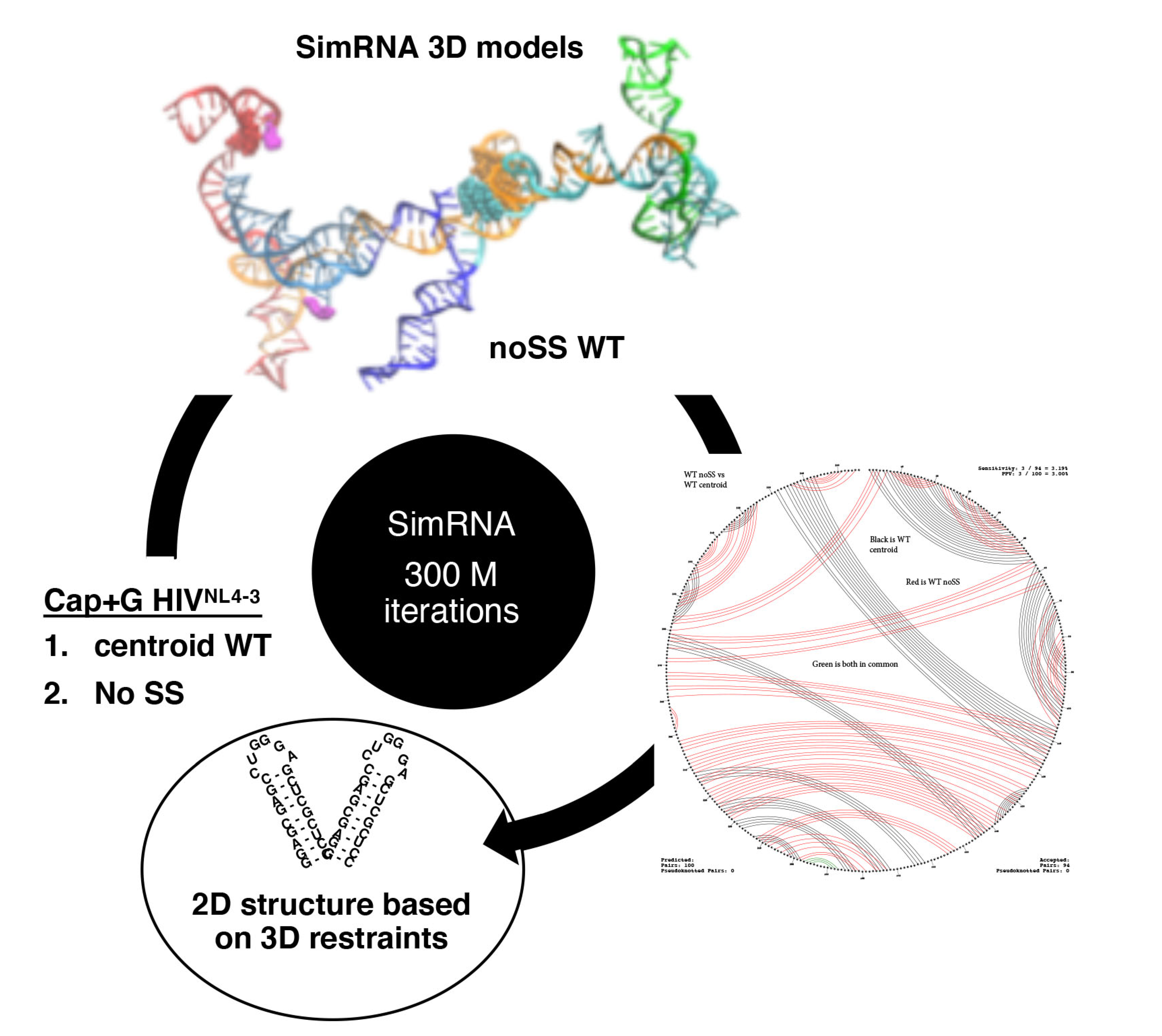
3.5. TAR Input 2D Restraints Significantly Influence SimRNA Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Westhof, E.; Fritsch, V. RNA folding: Beyond Watson–Crick pairs. Structure 2000, 8, R55–R65. [Google Scholar] [CrossRef]
- Westhof, E.; Masquida, B.; Jossinet, F. Predicting and Modeling RNA Architecture. Cold Spring Harb. Perspect. Biol. 2010, 3, a003632. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. RNA Structure: Advances and Assessment of 3D Structure Prediction. Annu. Rev. Biophys. 2017, 46, 483–503. [Google Scholar] [CrossRef]
- Lu, K.; Heng, X.; Garyu, L.; Monti, S.; Garcia, E.L.; Kharytonchyk, S.; Dorjsuren, B.; Kulandaivel, G.; Jones, S.; Hiremath, A.; et al. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 2011, 334, 242–245. [Google Scholar] [CrossRef] [PubMed]
- De Guzman, R.N.; Wu, Z.R.; Stalling, C.C.; Pappalardo, L.; Borer, P.N.; Summers, M.F. Structure of the HIV-1 Nucleocapsid Protein Bound to the SL3 -RNA Recognition Element. Science 1998, 279, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Kharytonchyk, S.; Chaudry, I.; Iyer, A.S.; Carter, H.; Becker, G.; Desai, Y.; Glang, L.; Choi, S.H.; Singh, K.; et al. Structural basis for transcriptional start site control of HIV-1 RNA fate. Science 2020, 368, 413–417. [Google Scholar] [PubMed]
- Berkhout, B. Structure and Function of the Human Immunodeficiency Virus Leader RNA. Prog. Nucleic Acid Res. Mol. Biol. 1996, 54, 1–34. [Google Scholar] [CrossRef]
- Huthoff, H.; Berkhout, B. Two alternating structures of the HIV-1 leader RNA. RNA 2001, 7, 143–157. [Google Scholar] [CrossRef]
- Keane, S.C.; Heng, X.; Lu, K.; Kharytonchyk, S.; Ramakrishnan, V.; Carter, G.; Barton, S.; Hosic, A.; Florwick, A.; Santos, J.; et al. Structure of the HIV-1 RNA packaging signal. Science 2015, 348, 917–921. [Google Scholar] [CrossRef]
- Abbink, T.E.M.; Ooms, M.; Haasnoot, P.C.J.; Berkhout, B. The HIV-1 Leader RNA Conformational Switch Regulates RNA Dimerization but Does Not Regulate mRNA Translation. Biochemistry 2005, 44, 9058–9066. [Google Scholar] [CrossRef]
- Smyth, R.P.; Negroni, M.; Lever, A.M.; Mak, J.; Kenyon, J.C. RNA Structure—A Neglected Puppet Master for the Evolution of Virus and Host Immunity. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.V.; Salguero, C.; Khan, S.N.; Meagher, J.L.; Brown, W.C.; Humbert, N.; De Rocquigny, H.; Smith, J.L.; D’Souza, V.M. HIV-1 Tat interactions with cellular 7SK and viral TAR RNAs identifies dual structural mimicry. Nat. Commun. 2018, 9, 4266. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Ela, F. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 1996, 24, 3974–3981. [Google Scholar] [CrossRef]
- Du, Z.; Lind, K.E.; James, T.L. Structure of TAR RNA Complexed with a Tat-TAR Interaction Nanomolar Inhibitor that Was Identified by Computational Screening. Chem. Biol. 2002, 9, 707–712. [Google Scholar] [CrossRef]
- Ennifar, E.; Walter, P.; Ehresmann, B.; Ehresmann, C.; Dumas, P. Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nat. Genet. 2001, 8, 1064–1068. [Google Scholar] [CrossRef]
- Baba, S.; Takahashi, K.-I.; Noguchi, S.; Takaku, H.; Koyanagi, Y.; Yamamoto, N.; Kawai, G. Solution RNA Structures of the HIV-1 Dimerization Initiation Site in the Kissing-Loop and Extended-Duplex Dimers. J. Biochem. 2005, 138, 583–592. [Google Scholar] [CrossRef]
- Renisio, J.-G.; Cosquer, S.; Cherrak, I.; El Antri, S.; Mauffret, O.; Fermandjian, S. Pre-organized structure of viral DNA at the binding-processing site of HIV-1 integrase. Nucleic Acids Res. 2005, 33, 1970–1981. [Google Scholar] [CrossRef]
- Lebars, I.; Legrand, P.; Aimé, A.; Pinaud, N.; Fribourg, S.; Di Primo, C. Exploring TAR–RNA aptamer loop–loop interaction by X-ray crystallography, UV spectroscopy and surface plasmon resonance. Nucleic Acids Res. 2008, 36, 7146–7156. [Google Scholar] [CrossRef]
- Ennifar, E.; Dumas, P. Polymorphism of Bulged-out Residues in HIV-1 RNA DIS Kissing Complex and Structure Comparison with Solution Studies. J. Mol. Biol. 2006, 356, 771–782. [Google Scholar] [CrossRef]
- Ferner, J.; Suhartono, M.; Breitung, S.; Jonker, H.R.A.; Hennig, M.; Wöhnert, J.; Göbel, M.; Schwalbe, H. Structures of HIV TAR RNA-Ligand Complexes Reveal Higher Binding Stoichiometries. ChemBioChem 2009, 10, 1490–1494. [Google Scholar] [CrossRef]
- Dubois, N.; Marquet, R.; Paillart, J.-C.; Bernacchi, S. Retroviral RNA Dimerization: From Structure to Functions. Front. Microbiol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Boeras, I.; Seufzer, B.; Brady, S.; Rendahl, A.; Heng, X.; Boris-Lawrie, K. The basal translation rate of authentic HIV-1 RNA is regulated by 5′UTR nt-pairings at junction of R and U5. Sci. Rep. 2017, 7, 6902. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.; MacKenzie, C.; Ayadi, L.; Lewin, S.R.; Branlant, C.; Purcell, D. Tat IRES modulator of tat mRNA (TIM-TAM): A conserved RNA structure that controls Tat expression and acts as a switch for HIV productive and latent infection. Nucleic Acids Res. 2019, 48, 2643–2660. [Google Scholar] [CrossRef]
- Nowakowski, J.; Tinoco, I. RNA Structure and Stability. Semin. Virol. 1997, 8, 153–165. [Google Scholar] [CrossRef]
- Brion, P.; Westhof, E. Hierarchy and Dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, I.; Bustamante, C. How RNA folds. J. Mol. Biol. 1999, 293, 271–281. [Google Scholar] [CrossRef]
- Batey, R.T.; Rambo, R.P.; Doudna, J.A. Tertiary Motifs in RNA Structure and Folding. Angew. Chem. Int. Ed. 1999, 38, 2326–2343. [Google Scholar] [CrossRef]
- Boeras, I.; Song, Z.; Moran, A.; Franklin, J.; Brown, W.C.; Johnson, M.; Boris-Lawrie, K.; Heng, X. DHX9/RHA Binding to the PBS-Segment of the Genomic RNA during HIV-1 Assembly Bolsters Virion Infectivity. J. Mol. Biol. 2016, 428, 2418–2429. [Google Scholar] [CrossRef]
- Zuker, M.; Sankoff, D. RNA secondary structures and their prediction. Bull. Math. Biol. 1984, 46, 591–621. [Google Scholar] [CrossRef]
- McCaskill, J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 1990, 29, 1105–1119. [Google Scholar] [CrossRef]
- Lu, Z.J.; Gloor, J.W.; Mathews, D.H. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA 2009, 15, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Boniecki, M.; Lach, G.; Dawson, W.K.; Tomala, K.; Lukasz, P.; Soltysiński, T.; Rother, K.M.; Bujnicki, J. SimRNA: A coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2015, 44, e63. [Google Scholar] [CrossRef]
- Magnus, M.; Boniecki, M.J.; Dawson, W.K.; Bujnicki, J. SimRNAweb: A web server for RNA 3D structure modeling with optional restraints. Nucleic Acids Res. 2016, 44, W315–W319. [Google Scholar] [CrossRef]
- Danhart, E.M.; Bakhtina, M.; Cantara, W.A.; Kuzmishin, A.B.; Ma, X.; Sanford, B.L.; Vargas-Rodriguez, O.; Košutić, M.; Goto, Y.; Suga, H.; et al. Conformational and chemical selection by a trans-acting editing domain. Proc. Natl. Acad. Sci. USA 2017, 114, E6774–E6783. [Google Scholar] [CrossRef]
- Brigham, B.S.; Kitzrow, J.P.; Reyes, J.-P.; Musier-Forsyth, K.; Munro, J.B. Intrinsic conformational dynamics of the HIV-1 genomic RNA 5′UTR. Proc. Natl. Acad. Sci. USA 2019, 116, 10372–10381. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Heng, X.; Kharytonchyk, S.; Garcia, E.L.; Lu, K.; Divakaruni, S.S.; Lacotti, C.; Edme, K.; Telesnitsky, A.; Summers, M.F. Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J. Mol. Biol. 2012, 417, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Abbink, T.E.M.; Berkhout, B. A Novel Long Distance Base-pairing Interaction in Human Immunodeficiency Virus Type 1 RNA Occludes the Gag Start Codon. J. Biol. Chem. 2002, 278, 11601–11611. [Google Scholar] [CrossRef]
- Mueller, N.; Van Bel, N.; Berkhout, B.; Das, A.T. HIV-1 splicing at the major splice donor site is restricted by RNA structure. Virology 2014, 468, 609–620. [Google Scholar] [CrossRef]
- Beerens, N.; Berkhout, B. The tRNA Primer Activation Signal in the Human Immunodeficiency Virus Type 1 Genome Is Important for Initiation and Processive Elongation of Reverse Transcription. J. Virol. 2002, 76, 2329–2339. [Google Scholar] [CrossRef]
- Jones, C.P.; Saadatmand, J.; Kleiman, L.; Musier-Forsyth, K. Molecular mimicry of human tRNALys anti-codon domain by HIV-1 RNA genome facilitates tRNA primer annealing. RNA 2012, 19, 219–229. [Google Scholar] [CrossRef]
- Laughrea, M.; Jetté, L. HIV-1 Genome Dimerization: Kissing-Loop Hairpin Dictates Whether Nucleotides Downstream of the 5′ Splice Junction Contribute to Loose and Tight Dimerization of Human Immunodeficiency Virus RNA. Biochemistry 1997, 36, 9501–9508. [Google Scholar] [CrossRef]
- Laughrea, M.; Shen, N.; Jetté, L.; Wainberg, M.A. Variant Effects of Non-Native Kissing-Loop Hairpin Palindromes on HIV Replication and HIV RNA Dimerization: Role of Stem−Loop B in HIV Replication and HIV RNA Dimerization. Biochemistry 1999, 38, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Silverman, R.H.; Jeang, K.-T. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 1989, 59, 273–282. [Google Scholar] [CrossRef]
- Berkhout, B.; Gatignol, A.; Silver, J.; Jeang, K.T. Efficient trans-activation by the HIV-2 tat protein requires a duplicated tar RNA structure. Nucleic Acids Res. 1990, 18, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Rounseville, M.P.; Kumar, A. Binding of a host cell nuclear protein to the stem region of human immunodeficiency virus type 1 trans-activation-responsive RNA. J. Virol. 1992, 66, 1688–1694. [Google Scholar] [CrossRef]
- Ippolito, J.A.; Steitz, T.A. A 1.3-A resolution crystal structure of the HIV-1 trans-activation response region RNA stem reveals a metal ion-dependent bulge conformation. Proc. Natl. Acad. Sci. USA 1998, 95, 9819–9824. [Google Scholar] [CrossRef]
- Das, A.T.; Klaver, B.; Berkhout, B. A Hairpin Structure in the R Region of the Human Immunodeficiency Virus Type 1 RNA Genome Is Instrumental in Polyadenylation Site Selection. J. Virol. 1999, 73, 81–91. [Google Scholar] [CrossRef]
- Van Bel, N.; Ghabri, A.; Das, A.T.; Berkhout, B. The HIV-1 leader RNA is exquisitely sensitive to structural changes. Virology 2015, 483, 236–252. [Google Scholar] [CrossRef]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA 2005, 11, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H. Revolutions in RNA Secondary Structure Prediction. J. Mol. Biol. 2006, 359, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Kharytonchyk, S.; Monti, S.; Smaldino, P.J.; Van, V.; Bolden, N.C.; Brown, J.D.; Russo, E.; Swanson, C.; Shuey, A.; Telesnitsky, A.; et al. Transcriptional start site heterogeneity modulates the structure and function of the HIV-1 genome. Proc. Natl. Acad. Sci. USA 2016, 113, 13378–13383. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.; Singh, G.; Bolinger, C.; Song, Z.; Boeras, I.; Weng, K.; Trent, B.; Brown, W.C.; Singh, K.; Boris-Lawrie, K.; et al. Virion-associated, host-derived DHX9/RNA helicase A enhances the processivity of HIV-1 reverse transcriptase on genomic RNA. J. Biol. Chem. 2019, 294, 11473–11485. [Google Scholar] [CrossRef]
- Bolinger, C.; Sharma, A.; Singh, D.; Yu, L.; Boris-Lawrie, K. RNA helicase A modulates translation of HIV-1 and infectivity of progeny virions. Nucleic Acids Res. 2010, 38, 1686–1696. [Google Scholar] [CrossRef]
- Sharma, A.; Yilmaz, A.; Marsh, K.; Cochrane, A.; Boris-Lawrie, K. Thriving under Stress: Selective Translation of HIV-1 Structural Protein mRNA during Vpr-Mediated Impairment of eIF4E Translation Activity. PLoS Pathog. 2012, 8, e1002612. [Google Scholar] [CrossRef]
- Singh, G.; Fritz, S.E.; Seufzer, B.; Boris-Lawrie, K. The mRNA encoding the JUND tumor suppressor detains nuclear RNA-binding proteins to assemble polysomes that are unaffected by mTOR. J. Biol. Chem. 2020, 295, 7763–7773. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Tran, T.; Vummidi, B.R.; Velagapudi, S.P.; Haniff, H.S.; Matsumoto, Y.; Crynen, G.; Southern, M.R.; Biswas, A.; Wang, Z.-F.; et al. A Massively Parallel Selection of Small Molecule-RNA Motif Binding Partners Informs Design of an Antiviral from Sequence. Chem 2018, 4, 2384–2404. [Google Scholar] [CrossRef]
- Patwardhan, N.N.; Ganser, L.R.; Kapral, G.J.; Eubanks, C.S.; Lee, J.; Sathyamoorthy, B.; Al-Hashimi, H.M.; Hargrove, A.E. Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. Medchemcomm 2017, 8, 1022–1036. [Google Scholar] [CrossRef]
- Wong, R.W.; Balachandran, A.; Cheung, P.K.; Cheng, R.; Pan, Q.; Stoilov, P.; Harrigan, P.R.; Blencowe, B.J.; Branch, D.R.; Cochrane, A. An activator of G protein-coupled receptor and MEK1/2-ERK1/2 signaling inhibits HIV-1 replication by altering viral RNA processing. PLoS Pathog. 2020, 16, e1008307. [Google Scholar] [CrossRef]
- Singh, G.; Rife, B.D.; Seufzer, B.; Salemi, M.; Rendahl, A.; Boris-Lawrie, K. Identification of conserved, primary sequence motifs that direct retrovirus RNA fate. Nucleic Acids Res. 2018, 46, 7366–7378. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solinska, J.; Shenoy, S.R.; Gareiss, P.; Krumpe, L.R.H.; Le Grice, S.F.J.; O’Keefe, B.R.; Schneekloth, J.J.S. Identification of Biologically Active, HIV TAR RNA-Binding Small Molecules Using Small Molecule Microarrays. J. Am. Chem. Soc. 2014, 136, 8402–8410. [Google Scholar] [CrossRef] [PubMed]
- Abulwerdi, F.A.; Shortridge, M.D.; Sztuba-Solinska, J.; Wilson, R.; Le Grice, S.F.J.; Varani, G.; Schneekloth, J.J.S. Development of Small Molecules with a Noncanonical Binding Mode to HIV-1 Trans Activation Response (TAR) RNA. J. Med. Chem. 2016, 59, 11148–11160. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, D.R.; Level, A.M.L. Antisense RNA sequences targeting the 5′ leader packaging signal region of human immunodeficiency virus type-1 inhibits viral replication at post-transcriptional stages of the life cycle. Gene Ther. 2000, 7, 1362–1368. [Google Scholar] [CrossRef][Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5′ Input Restraints a | Sensitivity b | PPV c |
|---|---|---|
| Dimer | 106/111 (96%) | 105/111 (95%) |
| Monomer | 73/99 (74%) | 73/99 (74%) |
| Mono Unpaired PolyA d | 77/99 (78%) | 77/89 (87%) |
| WT centroid | 94/94 (100%) | 94/123 (76%) |
| HIV NL4-3 5′-UTR 2D Restraints a | Sensitivity b | PPV c |
|---|---|---|
| Input | Versus Centroid WT Output | |
| Monomer | 54/94 (58%) | 54/99 (55%) |
| Dimer | 58/94 (62%) | 58/111 (52%) |
| noSS WT d | 3/100 (3%) | 3/94 (3%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osmer, P.S.; Singh, G.; Boris-Lawrie, K. A New Approach to 3D Modeling of Inhomogeneous Populations of Viral Regulatory RNA. Viruses 2020, 12, 1108. https://doi.org/10.3390/v12101108
Osmer PS, Singh G, Boris-Lawrie K. A New Approach to 3D Modeling of Inhomogeneous Populations of Viral Regulatory RNA. Viruses. 2020; 12(10):1108. https://doi.org/10.3390/v12101108
Chicago/Turabian StyleOsmer, Patrick S., Gatikrushna Singh, and Kathleen Boris-Lawrie. 2020. "A New Approach to 3D Modeling of Inhomogeneous Populations of Viral Regulatory RNA" Viruses 12, no. 10: 1108. https://doi.org/10.3390/v12101108
APA StyleOsmer, P. S., Singh, G., & Boris-Lawrie, K. (2020). A New Approach to 3D Modeling of Inhomogeneous Populations of Viral Regulatory RNA. Viruses, 12(10), 1108. https://doi.org/10.3390/v12101108