The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences




Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation to NGS Sequencing
2.1.1. Cells and Viruses
2.1.2. DNA Extraction and Preparation to NGS Sequencing
2.1.3. NGS Sequencing:
2.2. Molecular Characterization by Sanger Sequencing
2.2.1. Samples
2.2.2. Conventional PCR and Sequencing
2.3. Phylogenetic Analysis
2.3.1. Whole Genome Sequences
2.3.2. Sequences Obtained by Conventional Sequencing of O174L, K145R, MGF 505-5R and IGR I73R/I329L
2.4. Statistical Analysis
2.5. Mapping
3. Results
3.1. Next Generation Sequencing
3.2. Sanger Sequencing of Specific Regions
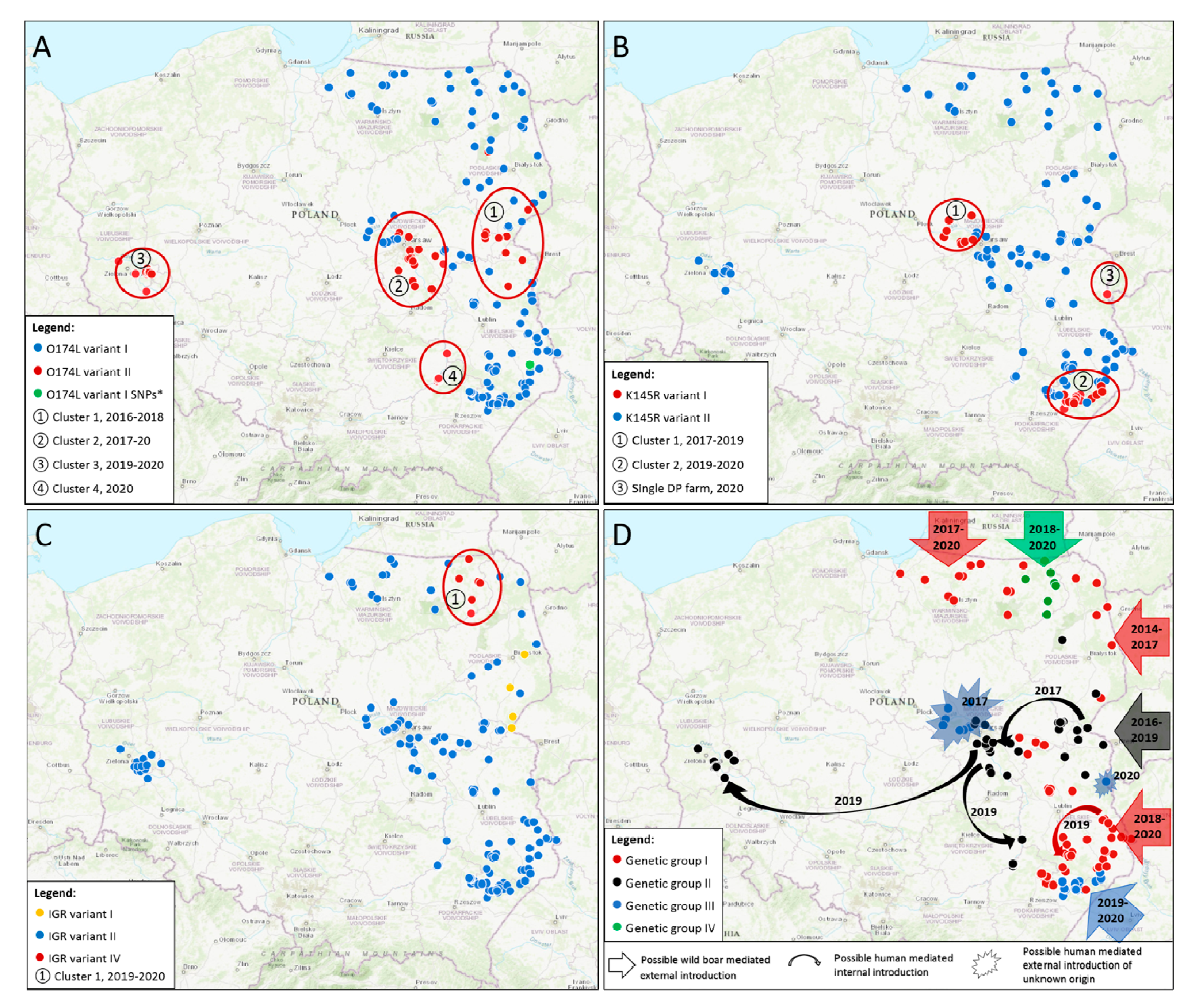
3.2.1. O174L Gene
3.2.2. K145R Gene
3.2.3. IGR I73R/I329L Region
3.2.4. MGF 505-5R Gene
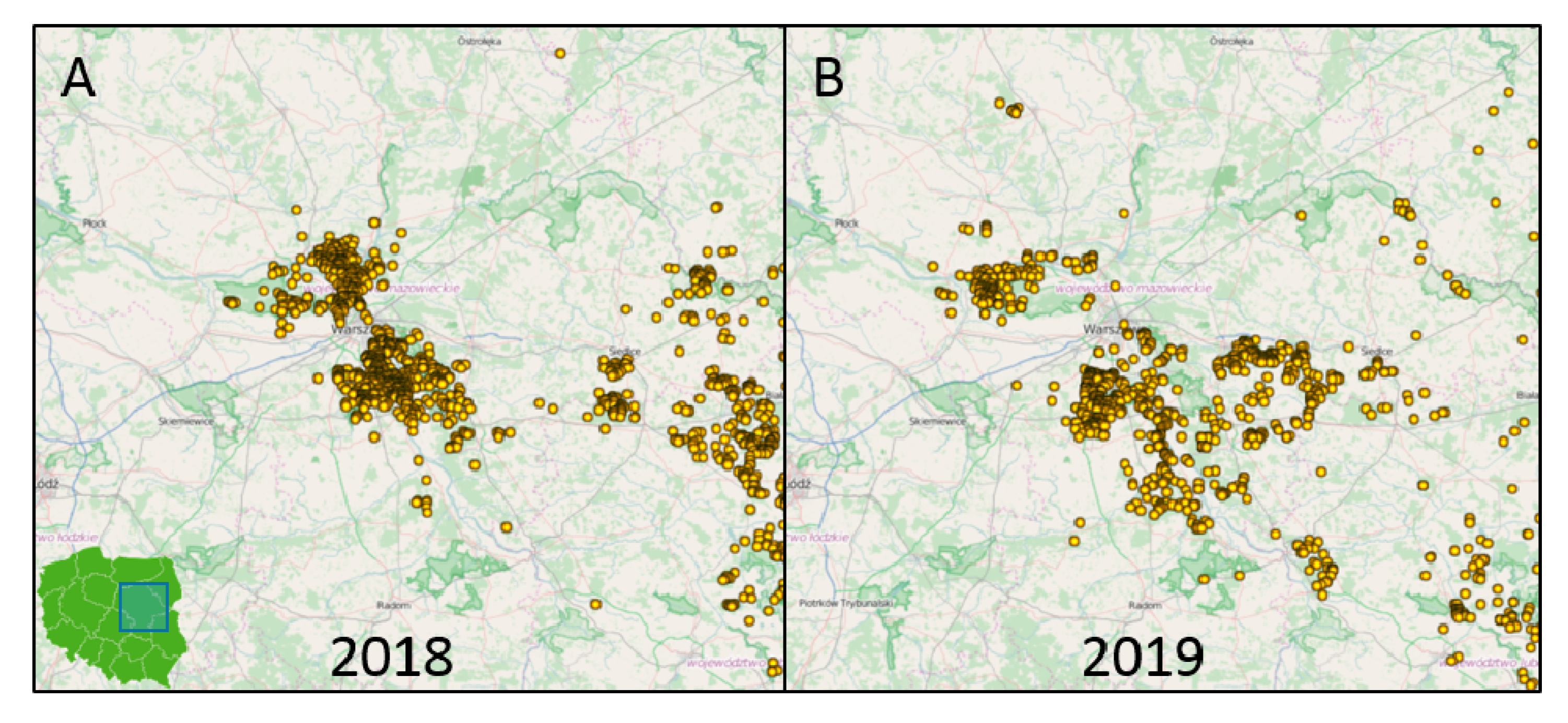
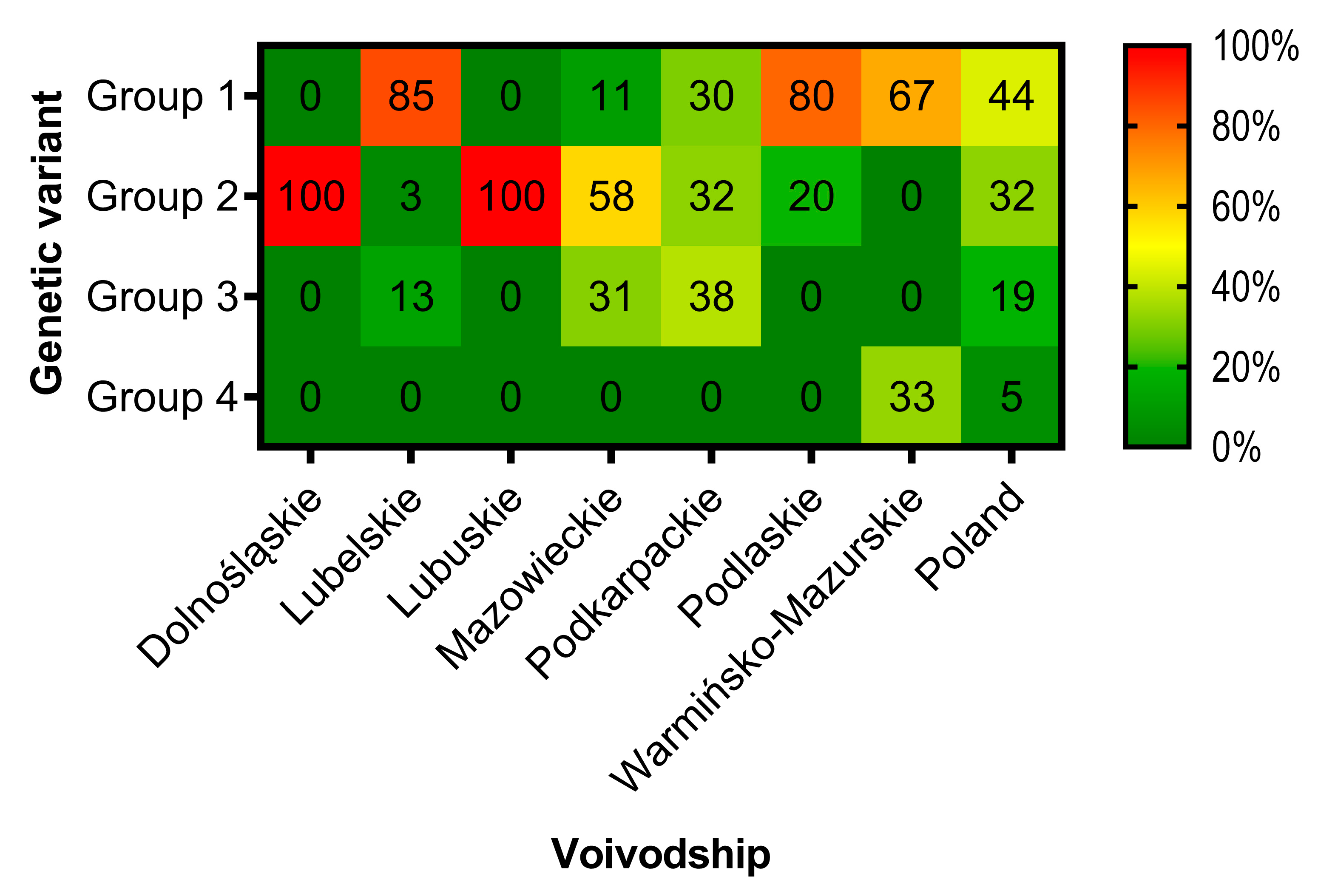
3.3. Phylogenetic Analysis and Genetic Groups Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Main Veterinary Inspectorate in Poland. Available online: https://www.wetgiw.gov.pl/main/komunikaty (accessed on 28 August 2020). (In Polish)
- Mazur-Panasiuk, N.; Woźniakowski, G.; Niemczuk, K. The first complete genomic sequences of African swine fever virus isolated in Poland. Sci. Rep. 2019, 9, 4556. [Google Scholar] [CrossRef] [PubMed]
- Olesen, A.S.; Lohse, L.; Dalgaard, M.D.; Woźniakowski, G.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Complete genome sequence of an African swine fever virus (ASFV POL/2015/Podlaskie) determined directly from pig erythrocyte-associated nucleic acid. J. Virol. Methods 2018, 261, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.; Forth, L.; King, J.; Groza, O.; Hübner, A.; Olesen, A.; Höper, D.; Dixon, L.; Netherton, C.; Rasmussen, T.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef]
- Olasz, F.; Mészáros, I.; Marton, S.; Kaján, G.L.; Tamás, V.; Locsmándi, G.; Magyar, T.; Bálint, Á.; Bányai, K.; Zádori, Z. A Simple Method for Sample Preparation to Facilitate Efficient Whole-Genome Sequencing of African Swine Fever Virus. Viruses 2019, 11, 1129. [Google Scholar] [CrossRef]
- Kovalenko, G.; Ducluzeau, A.-L.; Ishchenko, L.; Sushko, M.; Sapachova, M.; Rudova, N.; Solodiankin, O.; Gerilovych, A.; Dagdag, R.; Redlinger, M.; et al. Complete Genome Sequence of a Virulent African Swine Fever Virus from a Domestic Pig in Ukraine. Microbiol. Resour. Announc. 2019, 8, 17–19. [Google Scholar] [CrossRef]
- O’Donnell, V.K.; Grau, F.R.; Mayr, G.A.; Sturgill Samayoa, T.L.; Dodd, K.A.; Barrette, R.W. Rapid Sequence-Based Characterization of African Swine Fever Virus by Use of the Oxford Nanopore MinION Sequence Sensing Device and a Companion Analysis Software Tool. J. Clin. Microbiol. 2019, 58, 1–13. [Google Scholar] [CrossRef]
- Jia, L.; Jiang, M.; Wu, K.; Hu, J.; Wang, Y.; Quan, W.; Hao, M.; Liu, H.; Wei, H.; Fan, W.; et al. Nanopore sequencing of African swine fever virus. Sci. China Life Sci. 2020, 63, 160–164. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Woźniakowski, G. The unique genetic variation within the O174L gene of Polish strains of African swine fever virus facilitates tracking virus origin. Arch. Virol. 2019, 164, 1667–1672. [Google Scholar] [CrossRef]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic Variation among African Swine Fever Genotype II Viruses, Eastern and Central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef]
- Ge, S.; Liu, Y.; Li, L.; Wang, Q.; Li, J.; Ren, W.; Liu, C.; Bao, J.; Wu, X.; Wang, Z. An extra insertion of tandem repeat sequence in African swine fever virus, China, 2019. Virus Genes 2019, 55, 843–847. [Google Scholar] [CrossRef]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular Characterization of African Swine Fever Virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Ge, S.; Liu, Y.; Liu, C.; Liu, F.; Hu, Y.; Li, J.; Bao, J.; Ren, W.; et al. Infection of African swine fever in wild boar, China, 2018. Transbound. Emerg. Dis. 2019, 66, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Kolbasov, D.; Titov, I.; Tsybanov, S.; Gogin, A.; Malogolovkin, A. African Swine Fever Virus, Siberia, Russia, 2017. Emerg. Infect. Dis. 2018, 24, 796–798. [Google Scholar] [CrossRef]
- Frant, M.; Lyjak, M.; Bocian, L.; Barszcz, A.; Niemczuk, K.; Wozniakowski, G. African swine fever virus (ASFV) in Poland: Prevalence in a wild boar population (2017–2018). Vet. Med. 2020, 65, 143–158. [Google Scholar] [CrossRef]
- Main Veterinary Inspectorate in Poland—Current African Swine Fever Situaion in Poland (Map). Available online: https://bip.wetgiw.gov.pl/asf/mapa/ (accessed on 28 August 2020).
- Zhu, Z.; Xiao, C.-T.; Fan, Y.; Cai, Z.; Lu, C.; Zhang, G.; Jiang, T.; Tan, Y.; Peng, Y. Homologous recombination shapes the genetic diversity of African swine fever viruses. Vet. Microbiol. 2019, 236, 108380. [Google Scholar] [CrossRef] [PubMed]
- Michaud, V.; Randriamparany, T.; Albina, E. Comprehensive Phylogenetic Reconstructions of African Swine Fever Virus: Proposal for a New Classification and Molecular Dating of the Virus. PLoS ONE 2013, 8, e69662. [Google Scholar] [CrossRef] [PubMed]
- Quembo, C.J.; Jori, F.; Vosloo, W.; Heath, L. Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound. Emerg. Dis. 2018, 65, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Alkhamis, M.A.; Gallardo, C.; Jurado, C.; Soler, A.; Arias, M.; Sánchez-Vizcaíno, J.M. Phylodynamics and evolutionary epidemiology of African swine fever p72-CVR genes in Eurasia and Africa. PLoS ONE 2018, 13, e0192565. [Google Scholar] [CrossRef]
- Bastos, A.D.S.; Penrith, M.-L.; Crucière, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Gallardo, C.; Mwaengo, D.M.; Macharia, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martín, E.; Kasiti, J.; Bishop, R.P. Enhanced discrimination of African swine fever virus isolates through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef]
- Vilem, A.; Nurmoja, I.; Niine, T.; Riit, T.; Nieto, R.; Viltrop, A.; Gallardo, C. Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens 2020, 9, 582. [Google Scholar] [CrossRef]
- Gallardo, C.; Elsukova, A.; Woźniakowski, G.; Nieto, R.; Soler, A.; Sánchez-Vizcaíno, J.-M.; Igolkin, A.; Arias, M. Sequencing of the tandem repeat sequences (TRS) within the intergenic region between the multigene family 505 9R–10R genes: Additional tool for subtyping genotype II African swine fever (ASF) isolates. In Proceedings of the 11th International Congress for Veterinary Virology 12th Annual Meeting of EPIZONE, Vienna, Austria, 30 August 2018; p. 71. [Google Scholar]
- Zani, L.; Forth, J.H.; Forth, L.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Höper, D.; et al. Deletion at the 5′-end of Estonian ASFV strains associated with an attenuated phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wang, Q.; Lin, P.; Liu, C.; Li, L.; Wu, X.; Chi, T.; Xu, T.; Ge, S.; Liu, Y.; et al. Genome comparison of African swine fever virus China/2018/AnhuiXCGQ strain and related European p72 Genotype II strains. Transbound. Emerg. Dis. 2019, 66, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; He, X.; Zhang, X.; Zhang, X.; Liu, L.; Guan, Y.; Zhang, Y.; Bu, Z. Genome sequences derived from pig and dried blood pig feed samples provide important insights into the transmission of African swine fever virus in China in 2018. Emerg. Microbes Infect. 2019, 8, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Chenais, E.; Depner, K.; Guberti, V.; Dietze, K.; Viltrop, A.; Ståhl, K. Epidemiological considerations on African swine fever in Europe 2014–2018. Porc. Health Manag. 2019, 5, 6. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′->3′) | Position Referring to Georgia 2007/1 (FR682468.1) | Primer Length (nt) | Amplicon Length (nt) |
|---|---|---|---|---|
| O174L-F | TGGCTCAGACGATATTTCAACTC | 128,160–128,182 | 23 | 672 |
| O174L-R | GCCTCCACCACTTGAACCAT | 128,832–128,813 | 20 | |
| IGR-F | CTCAGAACTTTTTGAGAAGATTG | 172,236–172,258 | 23 | 349 |
| IGR-R | CAGCAAACAGTCCTATTGTT | 172,585–172,566 | 20 | |
| K145R-F | TTTCAGGCTGAAAACTTTTTAT | 65,030–65,051 | 22 | 282 |
| K145R-R | AAAGTTTTCAATGGTTGTTAGC | 65,312–65,291 | 22 | |
| MGF 505-5R-F | TACGCTTCTTTTCAATCATCAT | 37,968–37,989 | 22 | 598 |
| MGF 505-5R-R | AAATTAACAGTTGTTTGCCTTC | 38,608–38,587 | 22 |
| Genetic Group | O174L Variant | K145R Variant | IGR Variant | Occurrence (%) | Geographical Distribution |
|---|---|---|---|---|---|
| 0 | I | I | I | 0 | 100% similarity to Georgia 2007/1, generally absent * in Poland. |
| I | I | II | II | 43.51 | The most typical for Poland. |
| II | II | II | II | 32.47 | Southern Warsaw, western Poland, Tarnobrzeg clusters. |
| III | I | I | II | 19.48 | Northern Warsaw, southern Tomaszów Lubelski clusters. |
| IV | I | II | IV | 4.55 | Eastern Warrmińsko-Mazurskie cluster. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazur-Panasiuk, N.; Walczak, M.; Juszkiewicz, M.; Woźniakowski, G. The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses 2020, 12, 1094. https://doi.org/10.3390/v12101094
Mazur-Panasiuk N, Walczak M, Juszkiewicz M, Woźniakowski G. The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses. 2020; 12(10):1094. https://doi.org/10.3390/v12101094
Chicago/Turabian StyleMazur-Panasiuk, Natalia, Marek Walczak, Małgorzata Juszkiewicz, and Grzegorz Woźniakowski. 2020. "The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences" Viruses 12, no. 10: 1094. https://doi.org/10.3390/v12101094
APA StyleMazur-Panasiuk, N., Walczak, M., Juszkiewicz, M., & Woźniakowski, G. (2020). The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses, 12(10), 1094. https://doi.org/10.3390/v12101094