Distinct MCM10 Proteasomal Degradation Profiles by Primate Lentiviruses Vpr Proteins

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analysis and Vpr Alleles Preparation
2.2. Plasmid Construction
2.3. Cell Culture, Transfection, and Drug Treatment
2.4. Co-Immunoprecipitation Assay
2.5. Western Blotting
2.6. Immunofluorescence Staining
2.7. Cell Cycle Analysis
2.8. Real-Time qRT-PCR Analysis of Human MCM10 mRNA Expression
2.9. Statistical Analysis
3. Results
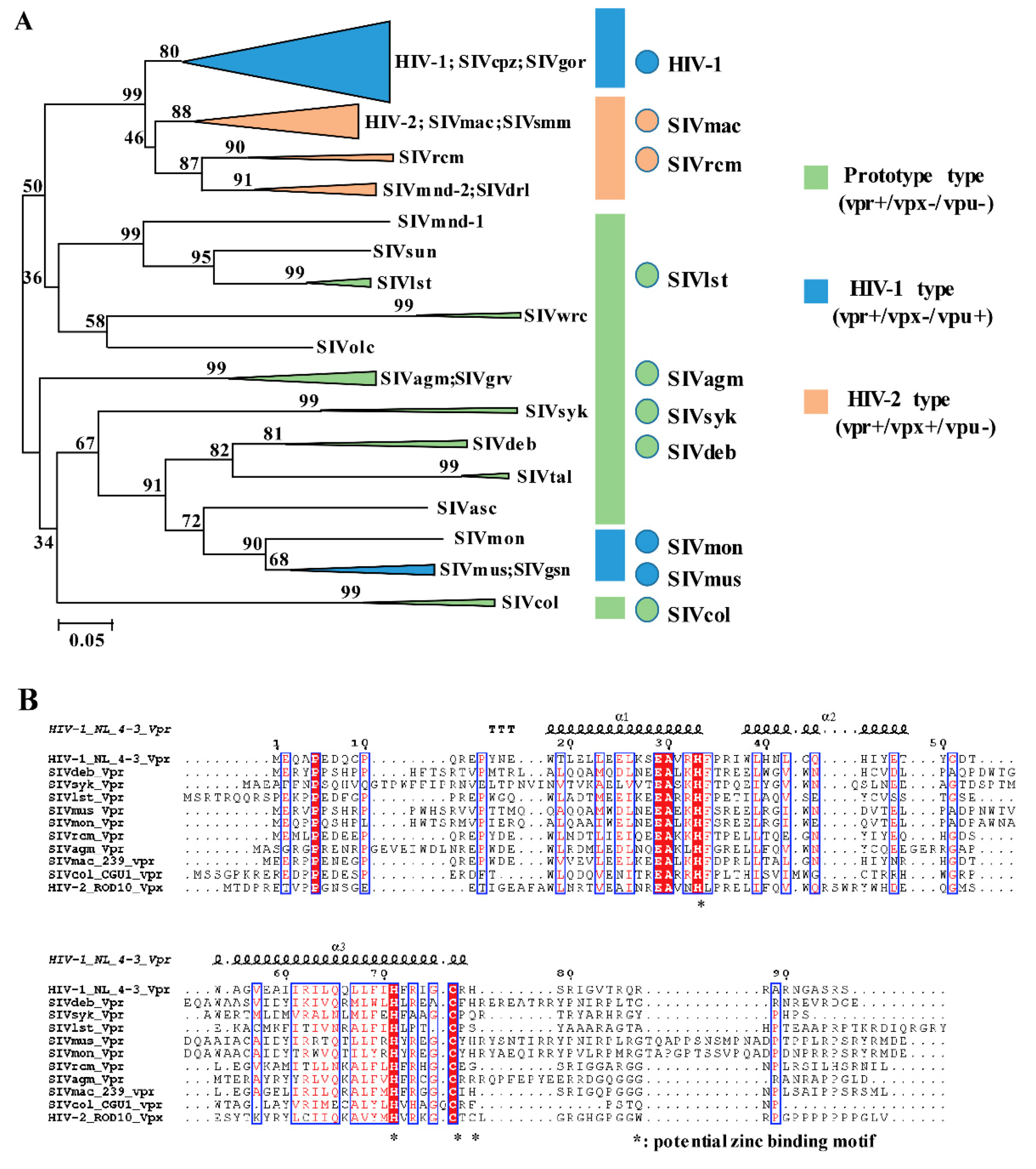
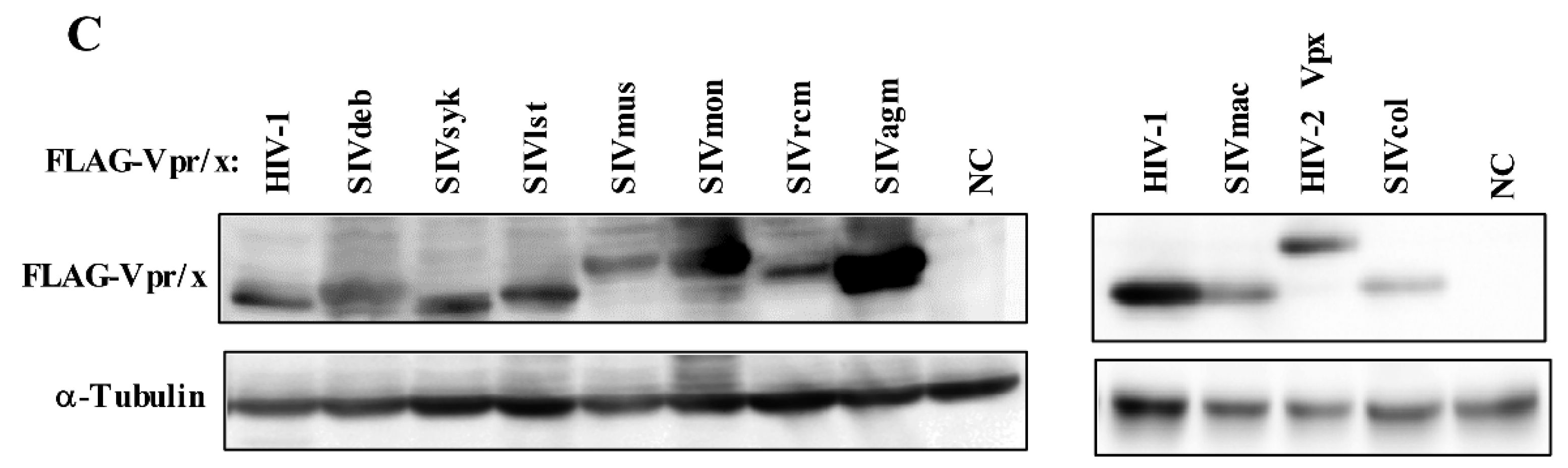
3.1. Phylogeny, Multiple Alignments, and Expression of Vpr/x from Representative Strains
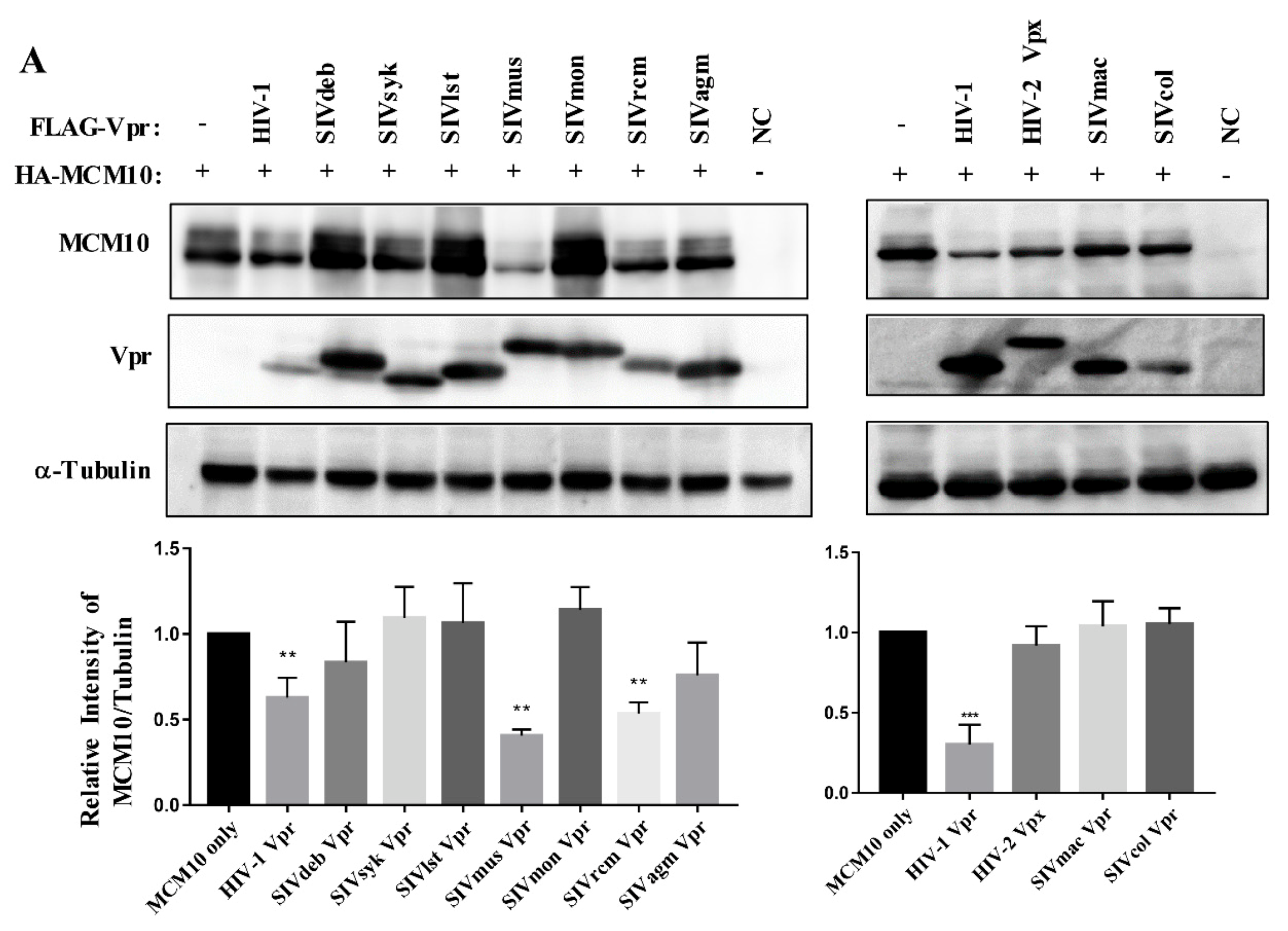
3.2. MCM10 down-Regulation by Primate Lentiviruses Vpr/x Proteins
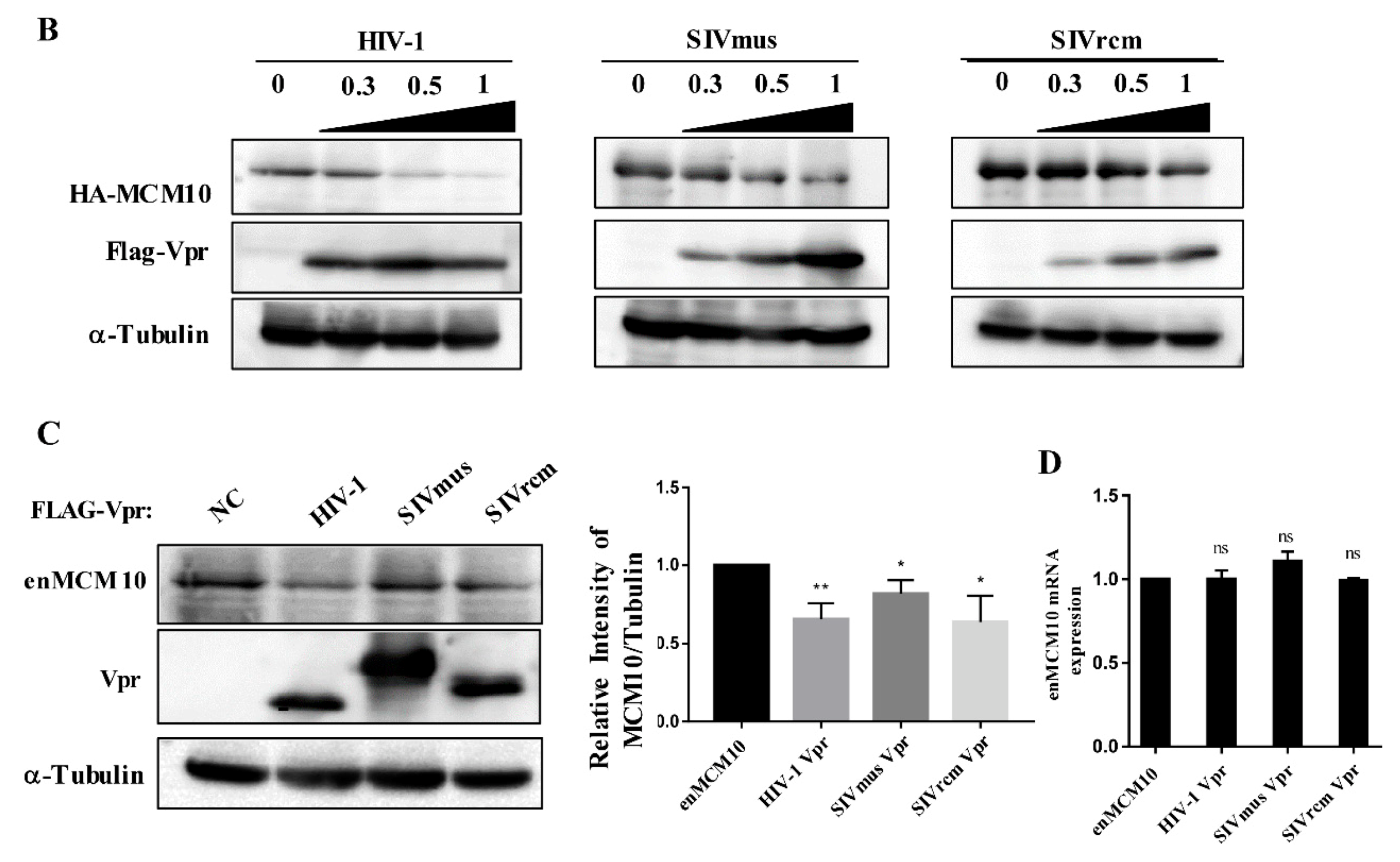
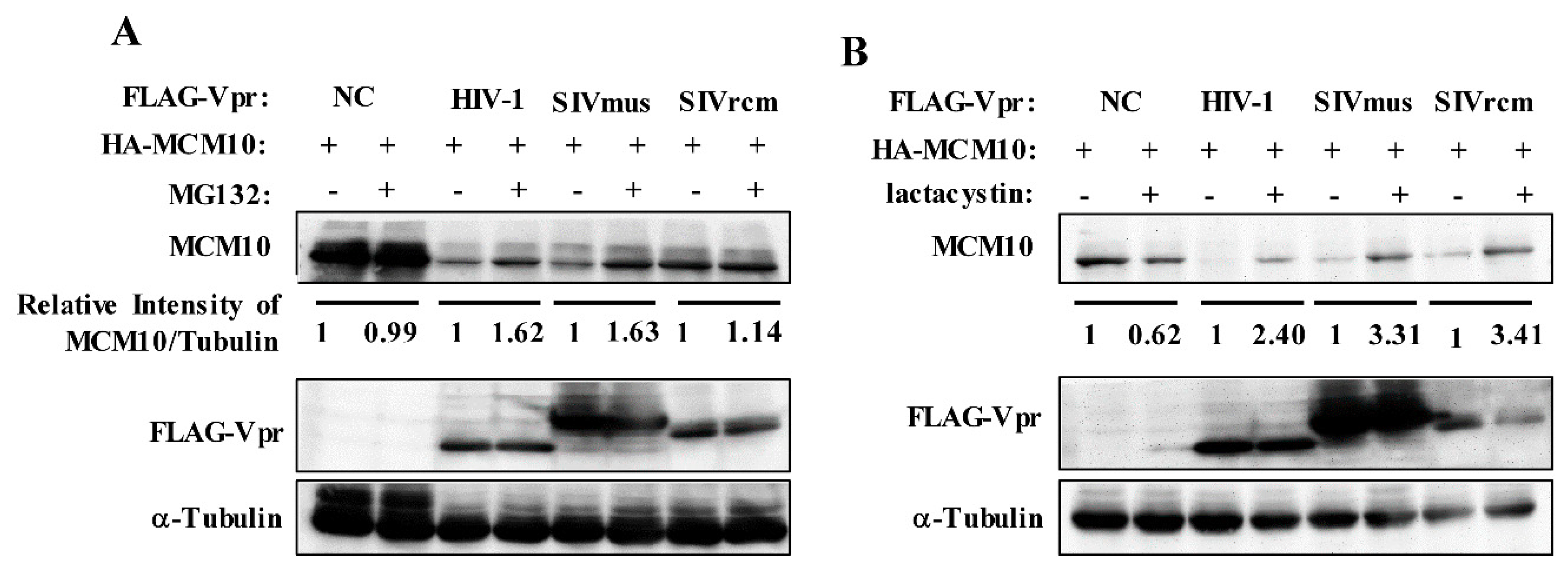
3.3. MCM10 Degradation via Proteasome Dependent Pathway
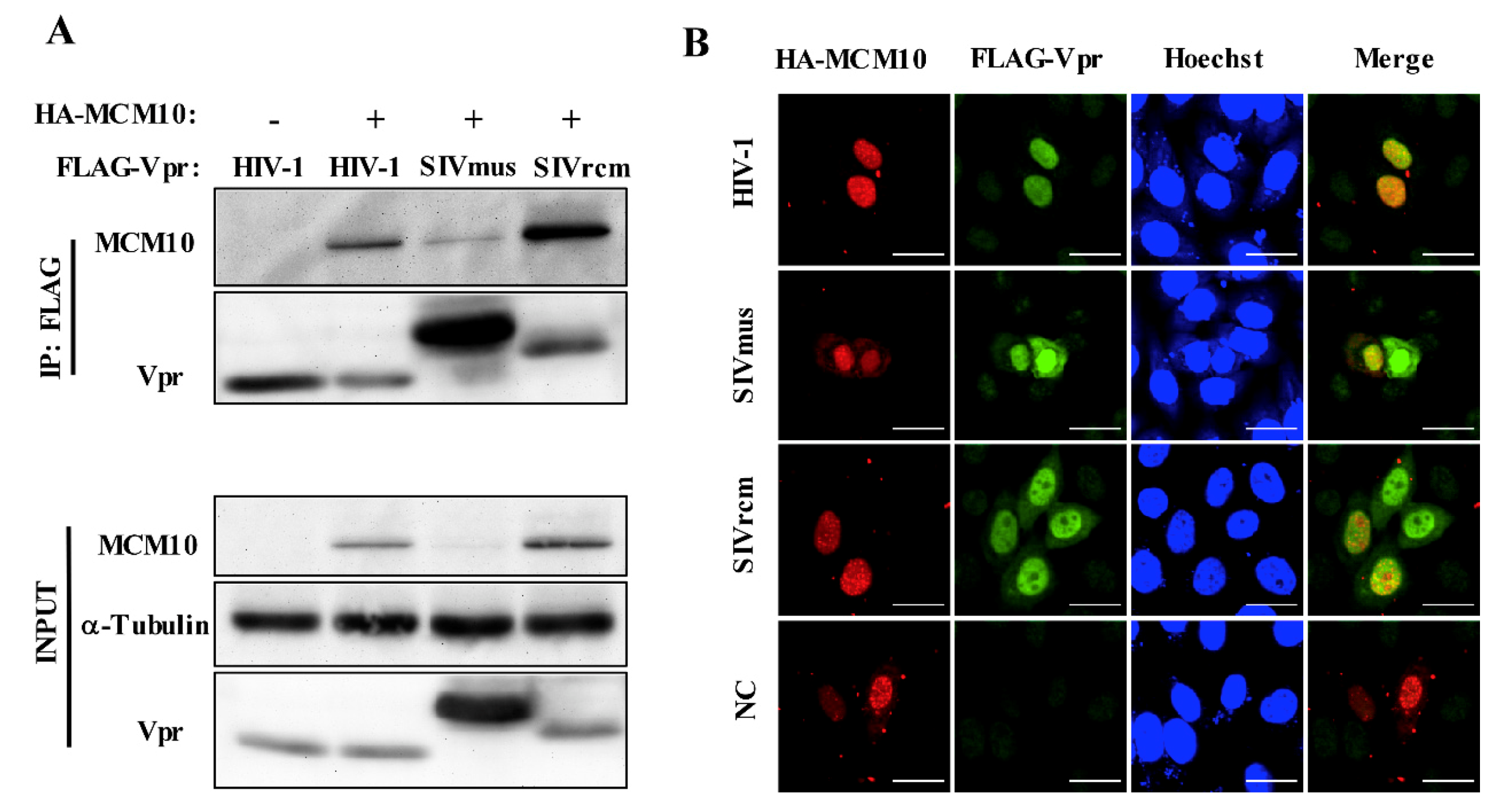
3.4. Interaction between MCM10 and HIV-1, SIVmus, and SIVrcm Vprs
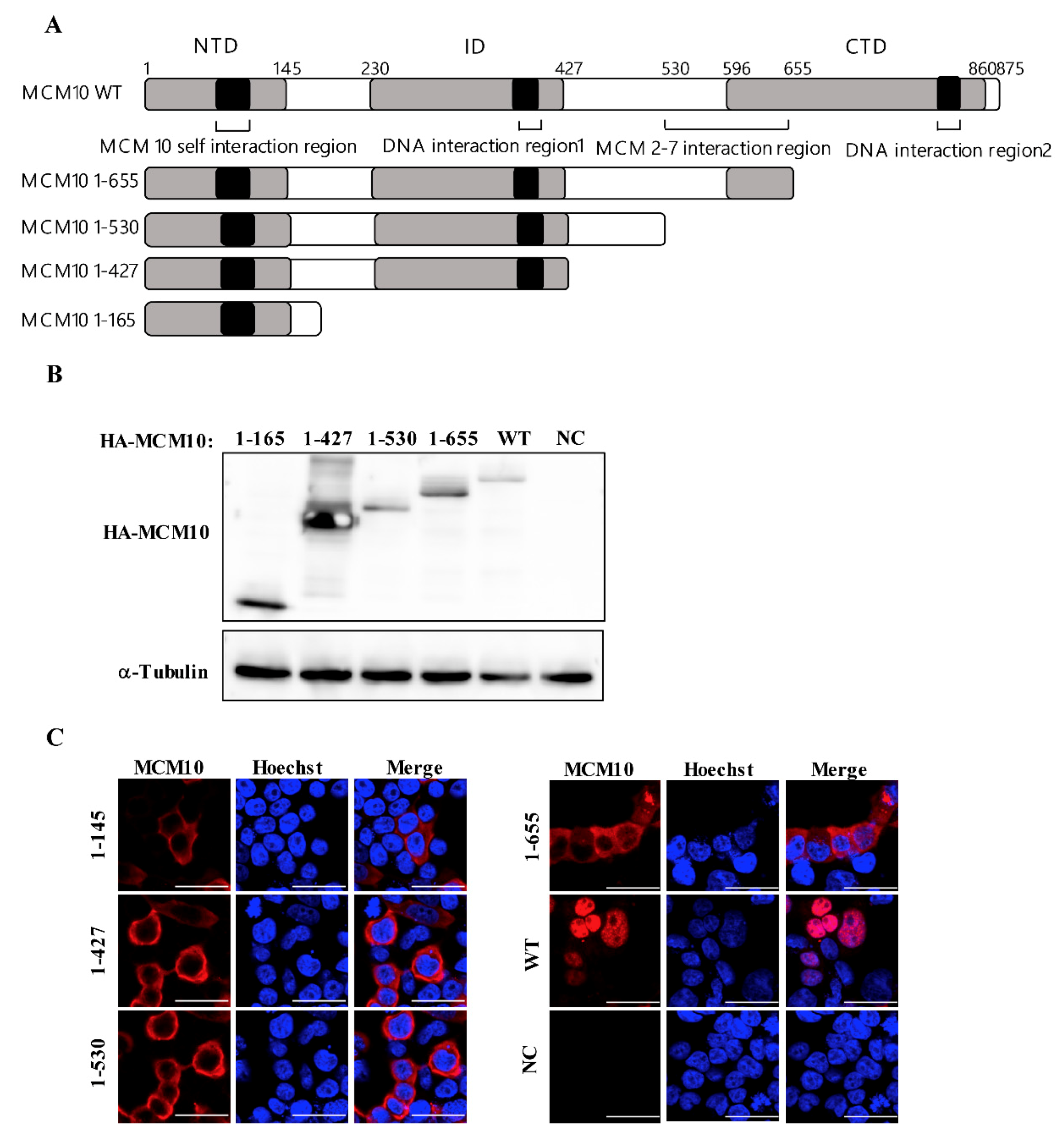
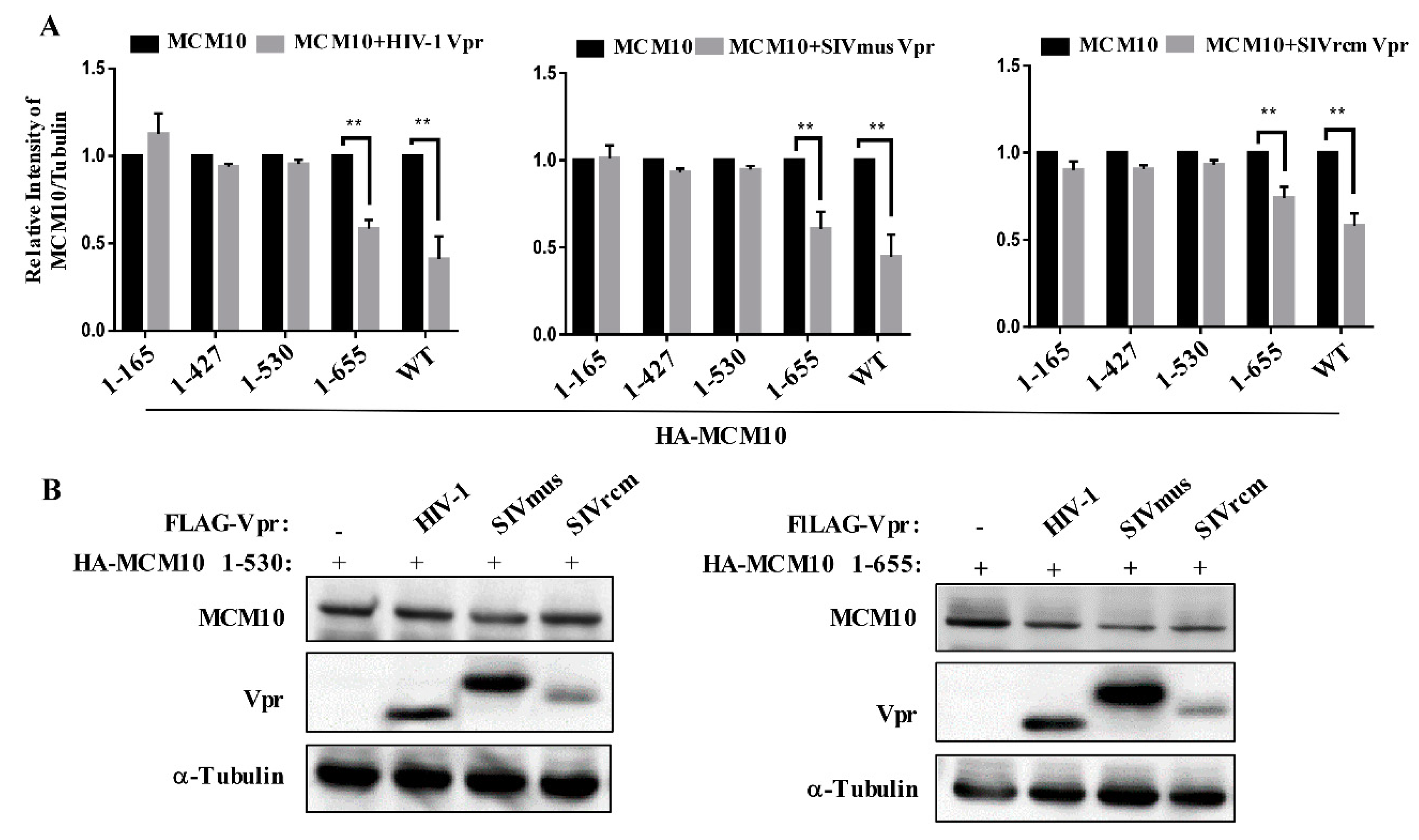
3.5. MCM 2-7 Interaction Region of MCM10 Susceptible to Degradation by Vprs
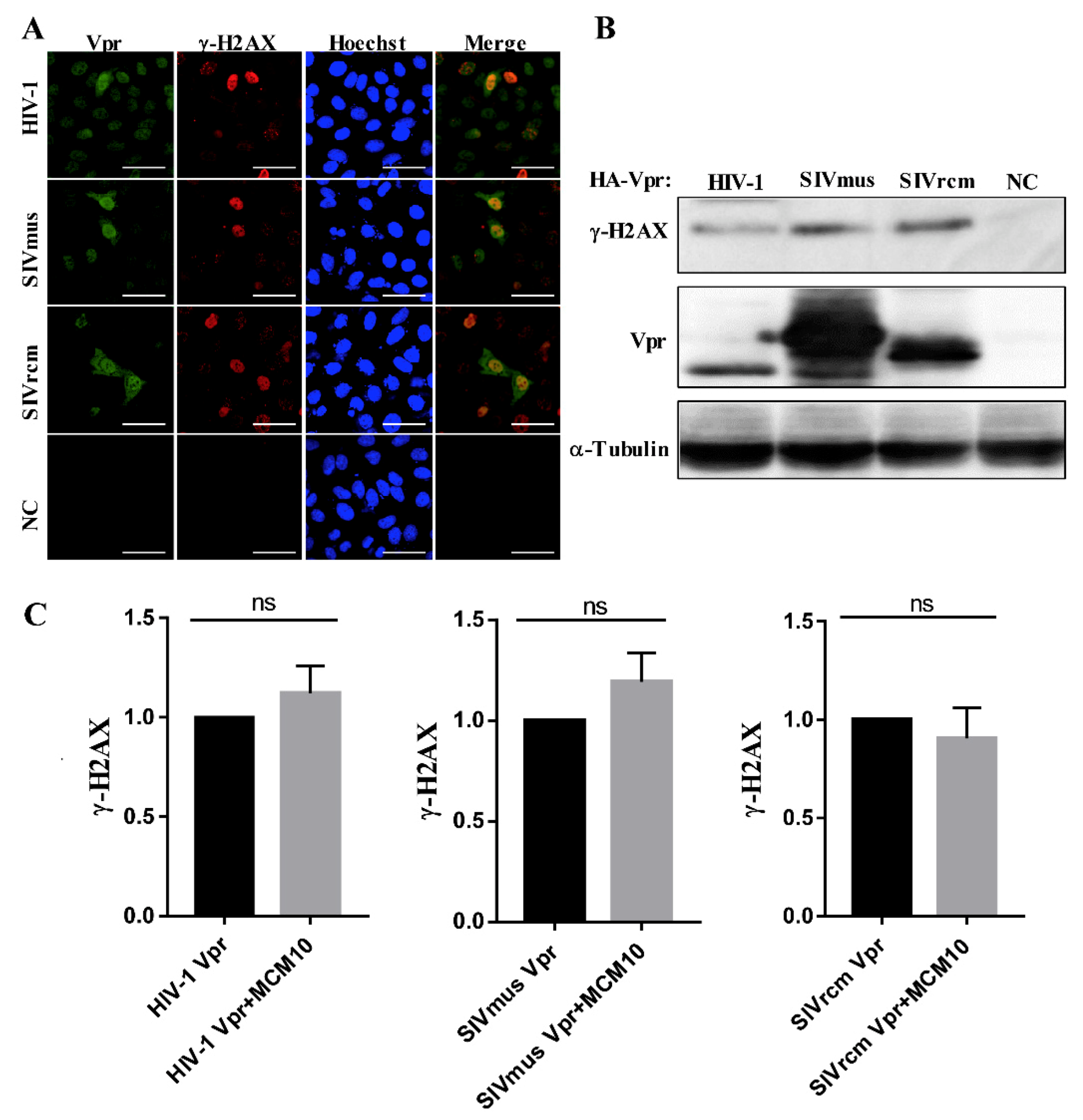
3.6. MCM10 Failure to Alleviate DDR Inducted by Primate Lentiviruses Vprs
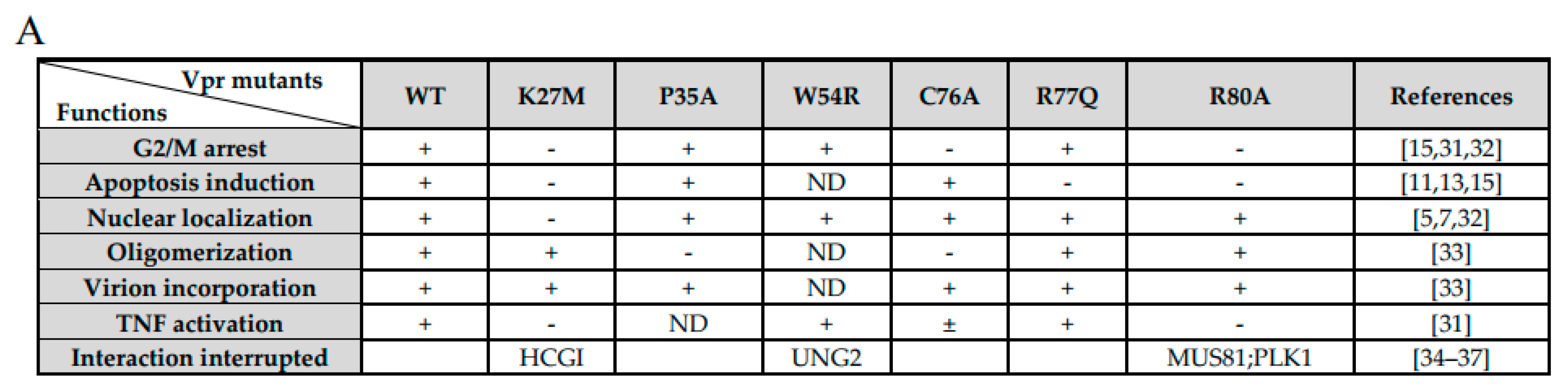
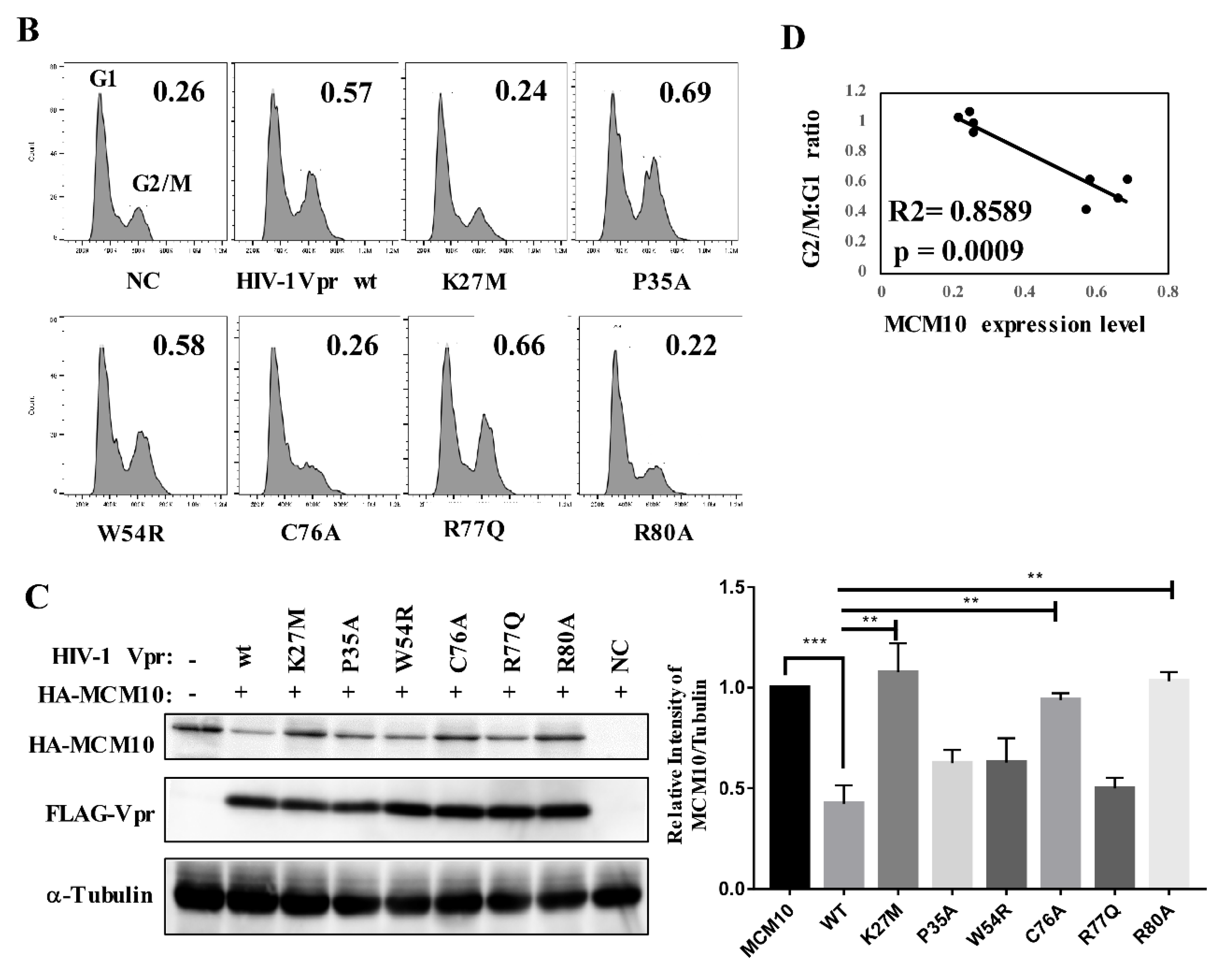
3.7. Correlation of MCM10 Degradation with HIV-1 Vpr G2/M Arrest
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Balliet, J.W.; Kolson, D.L.; Eiger, G.; Kim, F.M.; Mcgann, K.A.; Srinivasan, A.; Collman, R. Distinct Effects in Primary Macrophages and Lymphocytes of the Human-Immunodeficiency-Virus Type-1 Accessory Genes Vpr, Vpu, and Nef-Mutational Analysis of a Primary Hiv-1 Isolate. Virology 1994, 200, 623–631. [Google Scholar] [CrossRef]
- Mansky, L.M. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology 1996, 222, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Hay, R.T. Human immunodeficiency virus type 1 HIV-1 viral protein R Vpr interacts with Lys-tRNA synthetase: Implications for priming of HIV-1 reverse transcription. J. Virol. 1998, 72, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Heinzinger, N.K.; Bukrinsky, M.I.; Haggerty, S.A.; Ragland, A.M.; Kewalramani, V.; Lee, M.A.; Gendelman, H.E.; Ratner, L.; Stevenson, M.; Emerman, M. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. USA 1994, 91, 7311–7315. [Google Scholar] [CrossRef]
- Mahalingam, S.; Collman, R.G.; Patel, M.; Monken, C.E.; Srinivasan, A. Functional analysis of HIV-1 Vpr: Identification of determinants essential for subcellular localization. Virology 1995, 212, 331–339. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Kamata, M.; Yamamoto, T.; Zhang, X.; Miyamoto, Y.; Muneta, K.; Iijima, S.; Yoneda, Y.; Tsunetsugu-Yokota, Y.; Aida, Y. Novel nuclear import of Vpr promoted by importin alpha is crucial for human immunodeficiency virus type 1 replication in macrophages. J. Virol. 2007, 81, 5284–5293. [Google Scholar] [CrossRef]
- Felzien, L.K.; Woffendin, C.; Hottiger, M.O.; Subbramanian, R.A.; Cohen, E.A.; Nabel, G.J. HIV transcriptional activation by the accessory protein, VPR, is mediated by the p300 co-activator. Proc. Natl. Acad. Sci. USA 1998, 95, 5281–5286. [Google Scholar] [CrossRef]
- Kuramitsu, M.; Hashizume, C.; Yamamoto, N.; Azuma, A.; Kamata, M.; Yamamoto, N.; Tanaka, Y.; Aida, Y. A novel role for Vpr of human immunodeficiency virus type 1 as a regulator of the splicing of cellular pre-mRNA. Microbes Infect. 2005, 7, 1150–1160. [Google Scholar] [CrossRef]
- Hashizume, C.; Kuramitsu, M.; Zhang, X.F.; Kurosawa, T.; Kamata, M.; Aida, Y. Human immunodeficiency virus type 1 Vpr interacts with spliceosomal protein SAP145 to mediate cellular pre-mRNA splicing inhibition. Microbes Infect. 2007, 9, 490–497. [Google Scholar] [CrossRef]
- Rogel, M.E.; Wu, L.I.; Emerman, M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 1995, 69, 882–888. [Google Scholar] [CrossRef]
- Stewart, S.A.; Poon, B.; Jowett, J.B.; Chen, I.S. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 1997, 71, 5579–5592. [Google Scholar] [CrossRef]
- Nishizawa, M.; Kamata, M.; Mojin, T.; Nakai, Y.; Aida, Y. Induction of apoptosis by the Vpr protein of human immunodeficiency virus type 1 occurs independently of G2 arrest of the cell cycle. Virology 2000, 276, 16–26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fregoso, O.I.; Emerman, M. Activation of the DNA Damage Response Is a Conserved Function of HIV-1 and HIV-2 Vpr That Is Independent of SLX4 Recruitment. MBio 2016, 7, e01433-16. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Kim, B.; Jacquot, G.; Benichou, S.; Planelles, V. HIV-1 Vpr-induced apoptosis is cell cycle dependent and requires Bax but not ANT. PLoS Pathog. 2006, 2, e127. [Google Scholar] [CrossRef] [PubMed]
- Casey, L.; Wen, X.Y.; de Noronha, C.M.C. The functions of the HIV1 protein Vpr and its action through the DCAF1.DDB1. Cullin4 ubiquitin ligase. Cytokine 2010, 51, 1–9. [Google Scholar] [CrossRef]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature Activation of the SLX4 Complex by Vpr Promotes G2/M Arrest and Escape from Innate Immune Sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef]
- Romani, B.; Baygloo, N.S.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr Protein Enhances Proteasomal Degradation of MCM10 DNA Replication Factor through the Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Induce G2/M Cell Cycle Arrest. J. Biol. Chem. 2015, 290, 17380–17389. [Google Scholar] [CrossRef]
- Kaur, M.; Khan, M.M.; Kar, A.; Sharma, A.; Saxena, S. CRL4-DDB1-VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Res. 2012, 40, 7332–7346. [Google Scholar] [CrossRef]
- Thu, Y.M.; Bielinsky, A.K. MCM10: One tool for all-Integrity, maintenance and damage control. Semin. Cell Dev. Biol. 2014, 30, 121–130. [Google Scholar] [CrossRef]
- Baxley, R.M.; Bielinsky, A.K. Mcm10: A Dynamic Scaffold at Eukaryotic Replication Forks. Genes 2017, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Aida, Y. Visualizing Vpr-Induced G2 Arrest and Apoptosis. PLoS ONE 2014, 9, e86840. [Google Scholar] [CrossRef] [PubMed]
- Chutiwitoonchai, N.; Aida, Y. NXT1, a Novel Influenza A NP Binding Protein, Promotes the Nuclear Export of NP via a CRM1-Dependent Pathway. Viruses 2016, 8, 209. [Google Scholar] [CrossRef]
- Morellet, N.; Bouaziz, S.; Petitjean, P.; Roques, B.P. NMR structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 2003, 327, 215–227. [Google Scholar] [CrossRef]
- Miyatake, H.; Sanjoh, A.; Murakami, T.; Murakami, H.; Matsuda, G.; Hagiwara, K.; Yokoyama, M.; Sato, H.; Miyamoto, Y.; Dohmae, N.; et al. Molecular Mechanism of HIV-1 Vpr for Binding to Importin-alpha. J. Mol. Biol. 2016, 428, 2744–2757. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, X.H.; Barnes, C.O.; DeLucia, M.; Cohen, A.E.; Gronenborn, A.M.; Ahn, J.; Calero, G. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct. Mol. Biol. 2016, 23, 933–940. [Google Scholar] [CrossRef]
- Wang, H.; Guo, H.R.; Su, J.M.; Rui, Y.J.; Zheng, W.W.; Gao, W.Y.; Zhang, W.Y.; Li, Z.L.; Liu, G.C.; Markham, R.B.; et al. Inhibition of Vpx-Mediated SAMHD1 and Vpr-Mediated Host Helicase Transcription Factor Degradation by Selective Disruption of Viral CRL4 DCAF1 E3 Ubiquitin Ligase Assembly. J. Virol. 2017, 91, e00225-17. [Google Scholar] [CrossRef]
- Chang, H.; Siarot, L.; Murakami, T.; Aida, Y. Viral Infectious Diseases Unit, RIKEN, Wako, Saitama, Japan. Subcellular Distribution of Primate Lentiviruses 11 Vpr/x Proteins. Unpublished work. 2020. [Google Scholar]
- Chang, H.; Aida, Y. Viral Infectious Diseases Unit, RIKEN, Wako, Saitama, Japan. MCM10 Down-Regulation by Primate Lentiviruses 11 Vpr/x via Dose-Dependent Assay. Unpublished work. 2020. [Google Scholar]
- Roesch, F.; Richard, L.; Rua, R.; Porrot, F.; Casartelli, N.; Schwartz, O. Vpr Enhances Tumor Necrosis Factor Production by HIV-1-Infected T Cells. J. Virol. 2015, 89, 12118–12130. [Google Scholar] [CrossRef]
- Jacquot, G.; Le Rouzic, E.; David, A.; Mazzolini, J.; Bouchet, J.; Bouaziz, S.; Niedergang, F.; Pancino, G.; Benichou, S. Localization of HIV-1 Vpr to the nuclear envelope: Impact on Vpr functions and virus replication in macrophages. Retrovirology 2007, 4, 84. [Google Scholar] [CrossRef]
- Venkatachari, N.J.; Walker, L.A.; Tastan, O.; Le, T.; Dempsey, T.M.; Li, Y.; Yanamala, N.; Srinivasan, A.; Klein-Seetharaman, J.; Montelaro, R.C.; et al. Human immunodeficiency virus type 1 Vpr: Oligomerization is an essential feature for its incorporation into virus particles. Virol. J. 2010, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Preveral, S.; Selig, L.; Benarous, R.; Benichou, S. The interaction of Vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 in vivo mutation rate. J. Virol. 2000, 74, 7039–7047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.H.; DeLucia, M.; Ahn, J. SLX4-SLX1 Protein-independent Down-regulation of MUS81-EME1 Protein by HIV-1 Viral Protein R Vpr. J. Biol. Chem. 2016, 291, 16936–16947. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Blondot, M.L.; Chauveau, L.; Chougui, G.; Morel, M.; Leduc, M.; Guillonneau, F.; Ramirez, B.C.; Schwartz, O.; Margottin-Goguet, F. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 5311–5316. [Google Scholar] [CrossRef]
- Lv, L.; Wang, Q.; Xu, Y.P.; Tsao, L.C.; Nakagawa, T.; Guo, H.T.; Su, L.S.; Xiong, Y. Vpr Targets TET2 for Degradation by CRL4VprBP E3 Ligase to Sustain IL-6 Expression and Enhance HIV-1 Replication. Mol. Cell 2018, 70, 961–970. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef]
- Mehle, A.; Thomas, E.R.; Rajendran, K.S.; Gabuzda, D. A zinc-binding region in vif binds cul5 and determines cullin selection. J. Biol. Chem. 2006, 281, 17259–17265. [Google Scholar] [CrossRef]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef]
- Dannull, J.; Surovoy, A.; Jung, G.; Moelling, K. Specific Binding of Hiv-1 Nucleocapsid Protein to Psi-Rna in-Vitro Requires N-Terminal Zinc-Finger and Flanking Basic-Amino-Acid Residues. EMBO J. 1994, 13, 1525–1533. [Google Scholar] [CrossRef]
- Rein, A. RNA Packaging in HIV. Trends Microbiol. 2019, 27, 715–723. [Google Scholar] [CrossRef]
- Izumi, M.; Mizuno, T.; Yanagi, K.; Sugimura, K.; Okumura, K.; Imamoto, N.; Abe, T.; Hanaoka, F. The Mcm2-7-interacting domain of human mini-chromosome maintenance 10 Mcm10 protein is important for stable chromatin association and origin firing. J. Biol. Chem. 2017, 292, 13008–13021. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Warren, E.M.; Huang, H.; Fanning, E.; Chazin, W.J.; Eichman, B.F. Physical Interactions between Mcm10, DNA, and DNA Polymerase alpha. J. Biol. Chem. 2009, 284, 24662–24672. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Romani, B.; Baygloo, N.S.; Hamidi-Fard, M.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr Protein Induces Proteasomal Degradation of Chromatin-associated Class I HDACs to Overcome Latent Infection of Macrophages. J. Biol. Chem. 2016, 291, 2696–2711. [Google Scholar] [CrossRef]
- Yan, J.P.; Shun, M.C.; Hao, C.L.; Zhang, Y.; Qian, J.; Hrecka, K.; DeLucia, M.; Monnie, C.; Ahn, J.; Skowronski, J. HIV-1 Vpr Reprograms CLR4DCAF1 E3 Ubiquitin Ligase to Antagonize Exonuclease 1-Mediated Restriction of HIV-1 Infection. MBio 2018, 9, e01732-18. [Google Scholar] [CrossRef]
- Tomimatsu, N.; Mukherjee, B.; Harris, J.L.; Boffo, F.L.; Hardebeck, M.C.; Potts, P.R.; Khanna, K.K.; Burma, S. DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. J. Biol. Chem. 2017, 292, 10779–10790. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Bielinsky, A.K. Human Mcm10 regulates the catalytic subunit of DNA polymerase-alpha and prevents DNA damage during replication. Mol. Biol. Cell 2007, 18, 4085–4095. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.; Siarot, L.; Matsuura, R.; Lo, C.-W.; Sato, H.; Otsuki, H.; Aida, Y. Distinct MCM10 Proteasomal Degradation Profiles by Primate Lentiviruses Vpr Proteins. Viruses 2020, 12, 98. https://doi.org/10.3390/v12010098
Chang H, Siarot L, Matsuura R, Lo C-W, Sato H, Otsuki H, Aida Y. Distinct MCM10 Proteasomal Degradation Profiles by Primate Lentiviruses Vpr Proteins. Viruses. 2020; 12(1):98. https://doi.org/10.3390/v12010098
Chicago/Turabian StyleChang, Hao, Lowela Siarot, Ryosuke Matsuura, Chieh-Wen Lo, Hirotaka Sato, Hiroyuki Otsuki, and Yoko Aida. 2020. "Distinct MCM10 Proteasomal Degradation Profiles by Primate Lentiviruses Vpr Proteins" Viruses 12, no. 1: 98. https://doi.org/10.3390/v12010098
APA StyleChang, H., Siarot, L., Matsuura, R., Lo, C.-W., Sato, H., Otsuki, H., & Aida, Y. (2020). Distinct MCM10 Proteasomal Degradation Profiles by Primate Lentiviruses Vpr Proteins. Viruses, 12(1), 98. https://doi.org/10.3390/v12010098