When Dendritic Cells Go Viral: The Role of Siglec-1 in Host Defense and Dissemination of Enveloped Viruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
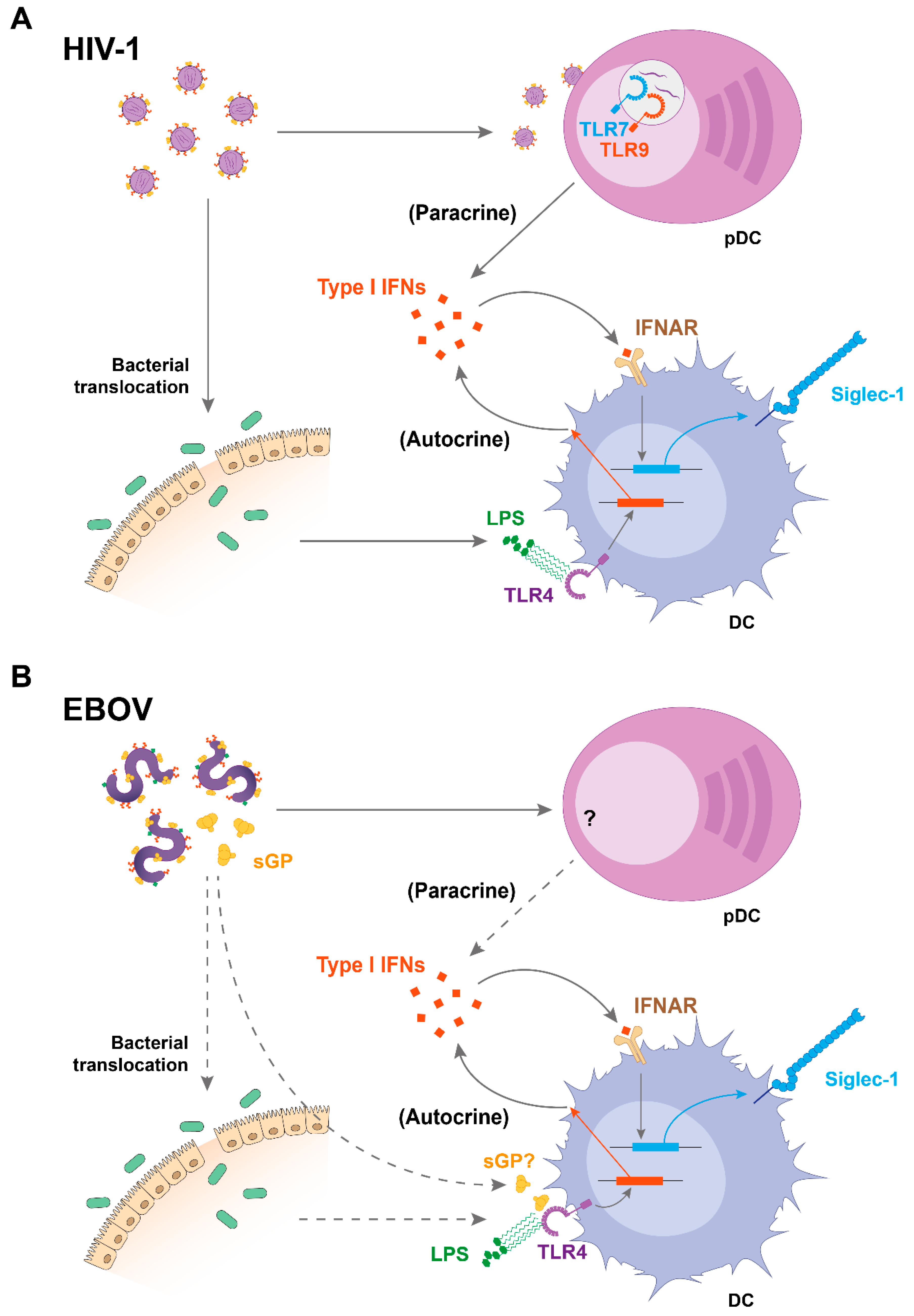
2. Viral Sensing and Immune Activation Triggers Siglec-1 Induction on DCs
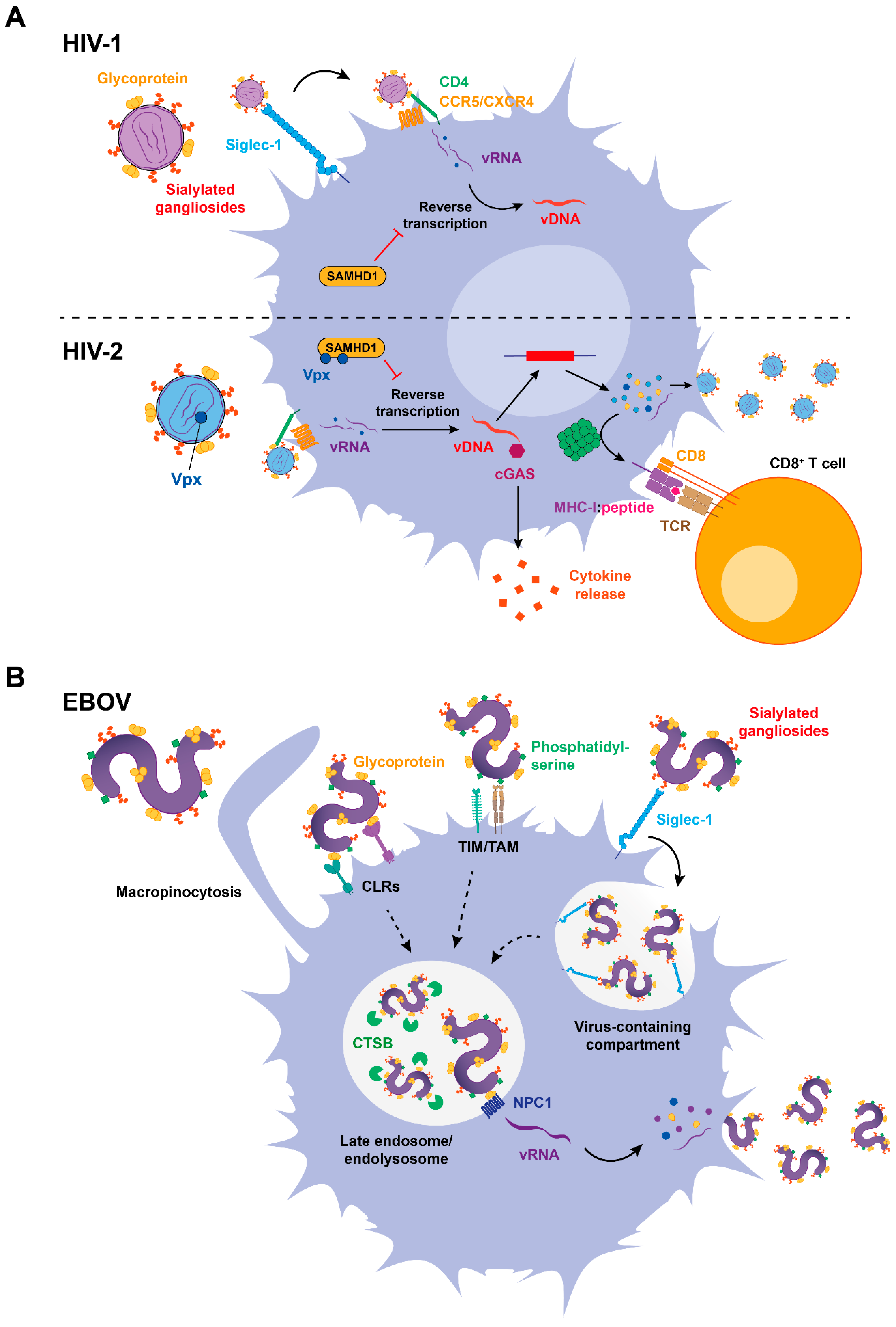
3. The Role of Siglec-1 in DC Infection and Antigen Presentation
4. Siglec-1 Captures Antigen-Containing Extracellular Vesicles, but This Mechanism also Promotes Viral Trans-Infection
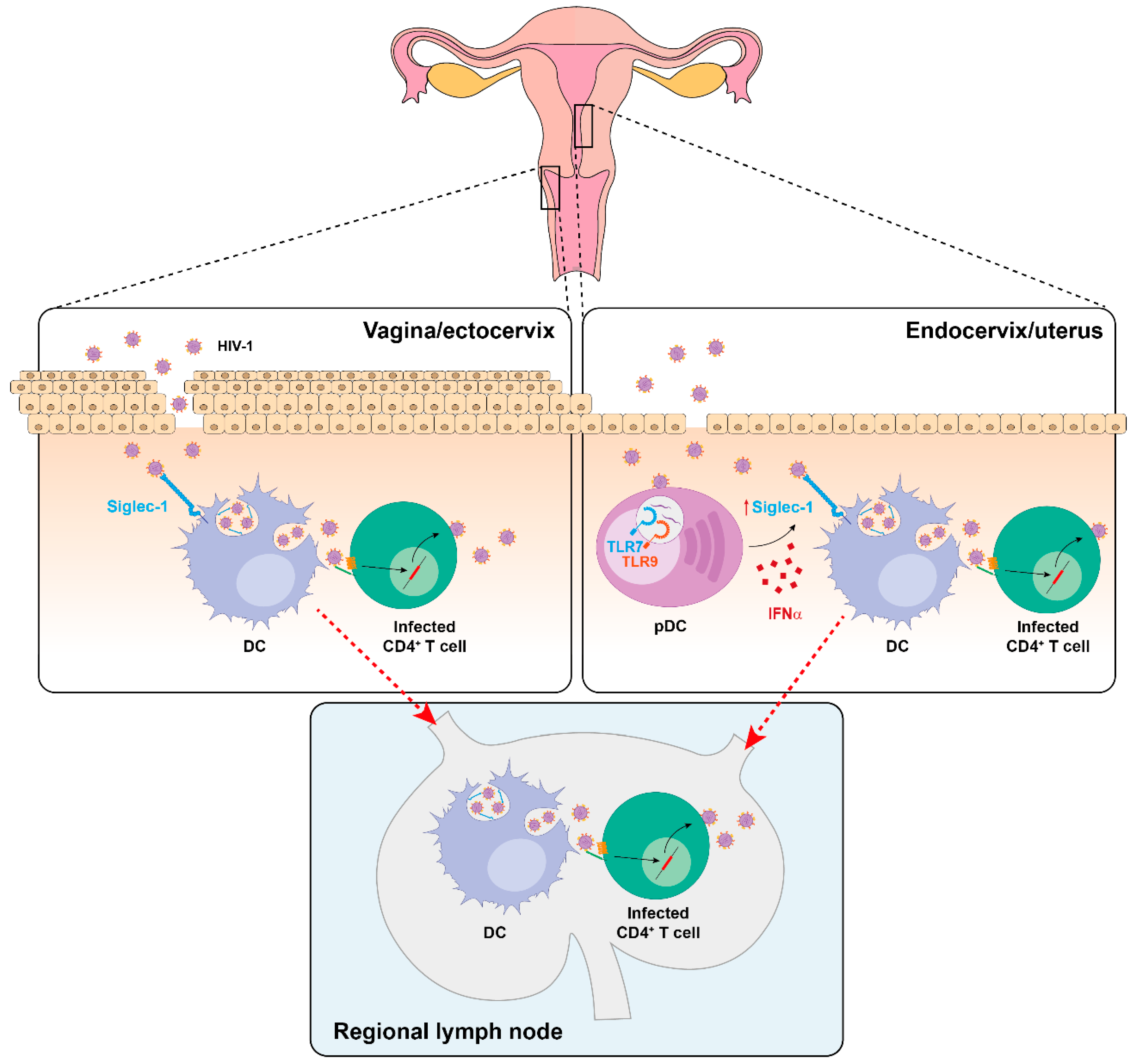
5. Siglec-1 Expression on Different Anatomical Compartments and Viral Dissemination Routes
6. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.C.; Malide, D.; Gibbs, J.S.; Bennink, J.R.; Yewdell, J.W. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 2002, 3, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science (80-) 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Wilson, N.S.; Behrens, G.M.N.; Lundie, R.J.; Smith, C.M.; Waithman, J.; Young, L.; Belz, G.T.; Carbone, F.R.; Crabb, B.S.; Health, W.R.; et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol. 2006, 7, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pollara, G.; Kwan, A.; Newton, P.J.; Handley, M.E.; Chain, B.M.; Katz, D.R. Dendritic cells in viral pathogenesis: Protective or defective? Int. J. Exp. Pathol. 2005, 86, 187–204. [Google Scholar] [CrossRef]
- Finlay, B.B.; McFadden, G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef]
- Rescigno, M. Dendritic cell functions: Learning from microbial evasion strategies. Semin. Immunol. 2015, 27, 119–124. [Google Scholar] [CrossRef]
- Cameron, P.U.; Freudenthal, P.S.; Barker, J.M.; Gezelter, S.; Inaba, K.; Steinman, R.M. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science (80-) 1992, 257, 383–387. [Google Scholar] [CrossRef]
- Knight, S.C.; Patterson, S. Bone marrow-derived dendritic cells, infection with human immunodeficiency virus, and immunopathology. Annu. Rev. Immunol. 1997, 15, 593–615. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: Evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar] [CrossRef]
- Spira, A.I.; Marx, P.A.; Patterson, B.K.; Mahoney, J.; Koup, R.A.; Wolinsky, S.M.; Ho, D.D. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J. Exp. Med. 1996, 183, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.-X.; Ma, L.; Liu, Q.-W.; Li, C.; Huang, Z.; Wu, L.; Xiong, S.-D.; Wang, J.-H.; Wang, J.-B. The molecule of DC-SIGN captures enterovirus 71 and confers dendritic cell-mediated viral trans-infection. Virol. J. 2014, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.A.W.P.; de Witte, L.; Bolmstedt, A.; van Kooyk, Y.; Geijtenbeek, T.B.H. Dendritic cells mediate herpes simplex virus infection and transmission through the C-type lectin DC-SIGN. J. Gen. Virol. 2008, 89, 2398–2409. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J. Dendritic cells as Achilles’ heel and Trojan horse during varicella zoster virus infection. Front. Microbiol. 2015, 6, 417. [Google Scholar] [PubMed]
- Farrell, H.E.; Bruce, K.; Lawler, C.; Oliveira, M.; Cardin, R.; Davis-Poynter, N.; Stevenson, P.G. Murine cytomegalovirus spreads by dendritic cell recirculation. MBio 2017, 8, e01264-17. [Google Scholar] [CrossRef] [PubMed]
- De Witte, L.; de Vries, R.D.; van der Vlist, M.; Yüksel, S.; Litjens, M.; de Swart, R.L.; Geijtenbeek, T.B.H. DC-SIGN and CD150 have distinct roles in transmission of measles virus from dendritic cells to T-lymphocytes. PLoS Pathog. 2008, 4, e1000049. [Google Scholar] [CrossRef]
- Pham, A.M.; Langlois, R.A.; TenOever, B.R. Replication in cells of hematopoietic origin is necessary for Dengue virus dissemination. PLoS Pathog. 2012, 8, e1002465. [Google Scholar] [CrossRef]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef]
- Izquierdo-Useros, N.; Lorizate, M.; McLaren, P.J.; Telenti, A.; Kräusslich, H.G.; Martinez-Picado, J. HIV-1 capture and transmission by dendritic cells: The role of viral glycolipids and the cellular receptor Siglec-1. PLoS Pathog. 2014, 10, e1004146. [Google Scholar] [CrossRef]
- Gummuluru, S.; Pina Ramirez, N.G.; Akiyama, H. CD169-dependent cell-associated HIV-1 transmission: A driver of virus dissemination. J. Infect. Dis. 2014, 210 (Suppl. S3), S641–S647. [Google Scholar] [CrossRef] [PubMed]
- Perez-Zsolt, D.; Erkizia, I.; Pino, M.; García-Gallo, M.; Martin, M.T.; Benet, S.; Chojnacki, J.; Fernández-Figueras, M.T.; Guerrero, D.; Urrea, V.; et al. Anti-Siglec-1 antibodies block Ebola viral uptake and decrease cytoplasmic viral entry. Nat. Microbiol. 2019, 4, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Hartnell, A.; Steel, J.; Turley, H.; Jones, M.; Jackson, D.G.; Crocker, P.R. Characterization of human sialoadhesin, a sialic acid binding receptor expressed by resident and inflammatory macrophage populations. Blood 2001, 97, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.; Uchil, P.D.; Jin, J.; Shui, G.; Ott, D.E.; Mothes, W.; Wenk, M.R. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J. Virol. 2008, 82, 11228–11238. [Google Scholar] [CrossRef]
- Lorizate, M.; Kräusslich, H.-G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef]
- Izquierdo-Useros, N.; Lorizate, M.; Contreras, F.-X.; Rodriguez-Plata, M.T.; Glass, B.; Erkizia, I.; Prado, J.G.; Casas, J.; Fabriàs, G.; Kräusslich, H.-G.; et al. Sialyllactose in viral membrane gangliosides is a novel molecular recognition pattern for mature dendritic cell capture of HIV-1. PLoS Biol. 2012, 10, e1001315. [Google Scholar] [CrossRef]
- Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.; Schmaljohn, A.; Aman, M.J. Lipid raft microdomains: A gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 2002, 195, 593–602. [Google Scholar] [CrossRef]
- Feizpour, A.; Yu, X.; Akiyama, H.; Miller, C.M.; Edmans, E.; Gummuluru, S.; Reinhard, B.M. Quantifying lipid contents in enveloped virus particles with plasmonic nanoparticles. Small 2015, 11, 1592–1602. [Google Scholar] [CrossRef][Green Version]
- Panchal, R.G.; Ruthel, G.; Kenny, T.A.; Kallstrom, G.H.; Lane, D.; Badie, S.S.; Li, L.; Bavari, S.; Aman, M.J. In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc. Natl. Acad. Sci. USA 2003, 100, 15936–15941. [Google Scholar] [CrossRef]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef]
- Munday, J.; Floyd, H.; Crocker, P.R. Sialic acid binding receptors (siglecs) expressed by macrophages. J. Leukoc Biol. 1999, 66, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Junt, T.; Moseman, E.A.; Iannacone, M.; Massberg, S.; Lang, P.A.; Boes, M.; Fink, K.; Henrickson, S.E.; Shayakhmetov, D.M.; Di Paolo, N.C.; et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 2007, 450, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Moseman, E.A.; Tonti, E.; Bosurgi, L.; Junt, T.; Henrickson, S.E.; Whelan, S.P.; Guidotti, L.G.; von Andrian, U.H. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature 2010, 465, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Ladinsky, M.S.; Uchil, P.D.; Beloor, J.; Pi, R.; Herrmann, C.; Motamedi, N.; Murooka, T.T.; Brehm, M.A.; Greiner, D.L.; et al. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science (80-) 2015, 350, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Uchil, P.D.; Pi, R.; Haugh, K.A.; Ladinsky, M.S.; Ventura, J.D.; Barrett, B.S.; Santiago, M.L.; Bjorkman, P.J.; Kassiotis, G.; Sewald, X.; et al. A protective role for the lectin CD169/Siglec-1 against a pathogenic murine retrovirus. Cell Host Microbe 2019, 25, 87–100. [Google Scholar] [CrossRef]
- UNAIDS Data Report (2019). Available online: https://www.unaids.org/en/resources/documents/2019/2019-UNAIDS-data (accessed on 16 December 2019).
- Baseler, L.; Chertow, D.S.; Johnson, K.M.; Feldmann, H.; Morens, D.M. The pathogenesis of Ebola virus disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 387–418. [Google Scholar] [CrossRef]
- Puryear, W.B.; Akiyama, H.; Geer, S.D.; Ramirez, N.P.; Yu, X.; Reinhard, B.M.; Gummuluru, S. Interferon-inducible mechanism of dendritic cell-mediated HIV-1 dissemination is dependent on Siglec-1/CD169. PLoS Pathog. 2013, 9, e1003291. [Google Scholar] [CrossRef]
- Rempel, H.; Calosing, C.; Sun, B.; Pulliam, L. Sialoadhesin expressed on IFN-induced monocytes binds HIV-1 and enhances infectivity. PLoS ONE 2008, 3, e1967. [Google Scholar] [CrossRef]
- Pino, M.; Erkizia, I.; Benet, S.; Erikson, E.; Fernández-Figueras, M.T.; Guerrero, D.; Dalmau, J.; Ouchi, D.; Rausell, A.; Ciuffi, A.; et al. HIV-1 immune activation induces Siglec-1 expression and enhances viral trans-infection in blood and tissue myeloid cells. Retrovirology 2015, 12, 37. [Google Scholar] [CrossRef]
- Von Sydow, M.; Sönnerborg, A.; Gaines, H.; Strannegård, Ö. Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res. Hum. Retroviruses 1991, 7, 375–380. [Google Scholar] [CrossRef]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.-H. The nature of the principal type 1 interferon-producing cells in human blood. Science (80-) 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Ferbas, J.J.; Toso, J.F.; Logar, A.J.; Navratil, J.S.; Rinaldo, C.R. CD4+ blood dendritic cells are potent producers of IFN-alpha in response to in vitro HIV-1 infection. J. Immunol. 1994, 152, 4649–4662. [Google Scholar] [PubMed]
- Yonezawa, A.; Morita, R.; Takaori-Kondo, A.; Kadowaki, N.; Kitawaki, T.; Hori, T.; Uchiyama, T. Natural alpha interferon-producing cells respond to human immunodeficiency virus type 1 with alpha interferon production and maturation into dendritic cells. J. Virol. 2003, 77, 3777–3784. [Google Scholar] [CrossRef]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.-J.; Hellman, J.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor Ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, A.; Louis, S.; Sourisseau, M.; Law, H.K.W.; Pothlichet, J.; Schilte, C.; Teigen, N.; Streeck, H.; Stellbrink, H.-J.; Hellman, J.; et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011, 7, e1001284. [Google Scholar] [CrossRef]
- Lehmann, C.; Harper, J.M.; Taubert, D.; Hartmann, P.; Fätkenheuer, G.; Jung, N.; van Lunzen, J.; Stellbrink, H.J.; Gallo, R.C.; Romerio, F. Increased interferon alpha expression in circulating plasmacytoid dendritic cells of HIV-1-infected patients. J. Acquir. Immune Defic. Syndr. 2008, 48, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Lafferty, M.; Garzino-Demo, A.; Jung, N.; Hartmann, P.; Fätkenheuer, G.; Wolf, J.S.; van Lunzen, J.; Romerio, F. Plasmacytoid dendritic cells accumulate and secrete interferon alpha in lymph nodes of HIV-1 patients. PLoS ONE 2010, 5, e11110. [Google Scholar] [CrossRef]
- Li, G.; Cheng, M.; Nunoya, J.; Cheng, L.; Guo, H.; Yu, H.; Liu, Y.; Su, L.; Zhang, L. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS Pathog. 2014, 10, e1004291. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.N.; Barry, A.P.; Vanderford, T.H.; Kozyr, N.; Chavan, R.; Klucking, S.; Barrat, F.J.; Coffman, R.L.; Staprans, S.I.; Feinberg, M.B. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat. Med. 2008, 14, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Fonteneau, J.-F.; Larsson, M.; Beignon, A.; Mckenna, K.; Dasilva, I.; Amara, A.; Liu, Y.; Lifson, J.D.; Littman, D.R.; Bhardwaj, N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 2004, 78, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Chang, J.J.; Chan, E.S.; Pollard, R.B.; Sidhu, H.K.; Kulkarni, S.; Wen, T.F.; Lindsay, R.J.; Orellana, L.; Mildvan, D.; et al. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat. Med. 2009, 15, 955–959. [Google Scholar] [CrossRef]
- Perez-Zsolt, D.; Cantero-Pérez, J.; Erkizia, I.; Benet, S.; Pino, M.; Serra-Peinado, C.; Hernández-Gallego, A.; Castellví, J.; Tapia, G.; Arnau-Saz, V.; et al. Dendritic cells from the cervical mucosa capture and transfer HIV-1 via Siglec-1. Front. Immunol. 2019, 10, 825. [Google Scholar] [CrossRef]
- Cella, M.; Salio, M.; Sakakibara, Y.; Langen, H.; Julkunen, I.; Lanzavecchia, A. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 1999, 189, 821–829. [Google Scholar] [CrossRef]
- Montoya, M.; Schiavoni, G.; Mattel, F.; Gresser, I.; Belardelli, F.; Borrow, P.; Tough, D.F. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002, 99, 3263–3271. [Google Scholar] [CrossRef]
- Gautier, G.; Humbert, M.; Deauvieau, F.; Scuiller, M.; Hiscott, J.; Bates, E.E.M.; Trinchieri, G.; Caux, C.; Garrone, P. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J. Exp. Med. 2005, 201, 1435–1446. [Google Scholar] [CrossRef]
- Pollara, G.; Handley, M.E.; Kwan, A.; Chain, B.M.; Katz, D.R. Autocrine type I interferon amplifies dendritic cell responses to lipopolysaccharide via the nuclear factor-κB/p38 pathways. Scand. J. Immunol. 2006, 63, 151–154. [Google Scholar] [CrossRef]
- Estes, J.D.; Harris, L.D.; Klatt, N.R.; Tabb, B.; Pittaluga, S.; Paiardini, M.; Barclay, G.R.; Smedley, J.; Pung, R.; Oliveira, K.M.; et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010, 6, e1001052. [Google Scholar] [CrossRef]
- Hensley-McBain, T.; Berard, A.R.; Manuzak, J.A.; Miller, C.J.; Zevin, A.S.; Polacino, P.; Gile, J.; Agricola, B.; Cameron, M.; Hu, S.-L.; et al. Intestinal damage precedes mucosal immune dysfunction in SIV infection. Nat. Mucosal. Immunol. 2018, 11, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Useros, N.; Lorizate, M.; Puertas, M.C.; Rodriguez-Plata, M.T.; Zangger, N.; Erikson, E.; Pino, M.; Erkizia, I.; Glass, B.; Clotet, B.; et al. Siglec-1 is a novel dendritic cell receptor that mediates HIV-1 trans-infection through recognition of viral membrane gangliosides. PLoS Biol. 2012, 10, e1001448. [Google Scholar] [CrossRef] [PubMed]
- Villinger, F.; Rollin, P.E.; Brar, S.S.; Chikkala, N.F.; Winter, J.; Sundstrom, J.B.; Zaki, S.R.; Swanepoel, R.; Ansari, A.A.; Peters, C.J. Markedly elevated levels of interferon (IFN)-γ, IFN-α, interleukin (IL)-2, IL-10, and tumor necrosis factor-α associated with fatal Ebola virus infection. J. Infect. Dis. 1999, 179 (Suppl. S1), S188–S191. [Google Scholar] [CrossRef] [PubMed]
- Caballero, I.S.; Honko, A.N.; Gire, S.K.; Winnicki, S.M.; Melé, M.; Gerhardinger, C.; Lin, A.E.; Rinn, J.L.; Sabeti, P.C.; Hensley, L.E.; et al. In vivo Ebola virus infection leads to a strong innate response in circulating immune cells. BMC Genomics 2016, 17, 707. [Google Scholar] [CrossRef]
- Leroy, E.M.; Baize, S.; Volchkov, V.E.; Fisher-Hoch, S.P.; Georges-Courbot, M.C.; Lansoud-Soukate, J.; Capron, M.; Debré, P.; McCormick, J.B.; Georges, A.J. Human asymptomatic Ebola infection and strong inflammatory response. Lancet 2000, 355, 2210–2215. [Google Scholar] [CrossRef]
- Leung, L.W.; Martinez, O.; Reynard, O.; Volchkov, V.E.; Basler, C.F. Ebola virus failure to stimulate plasmacytoid dendritic cell interferon responses correlates with impaired cellular entry. J. Infect. Dis. 2011, 204 (Suppl. S3), S973–S977. [Google Scholar] [CrossRef]
- Versteeg, K.; Menicucci, A.R.; Woolsey, C.; Mire, C.E.; Geisbert, J.B.; Cross, R.W.; Agans, K.N.; Jeske, D.; Messaoudi, I.; Geisbert, T.W. Infection with the Makona variant results in a delayed and distinct host immune response compared to previous Ebola virus variants. Sci. Rep. 2017, 7, 9370. [Google Scholar] [CrossRef]
- Ayithan, N.; Bradfute, S.B.; Anthony, S.M.; Stuthman, K.S.; Dye, J.M.; Bavari, S.; Bray, M.; Ozato, K. Ebola virus-like particles stimulate type I interferons and proinflammatory cytokine expression through the Toll-like receptor and interferon signaling pathways. J. Interf. Cytokine Res. 2014, 34, 79–89. [Google Scholar] [CrossRef]
- Olejnik, J.; Forero, A.; Deflubé, L.R.; Hume, A.J.; Manhart, W.A.; Nishida, A.; Marzi, A.; Katze, M.G.; Ebihara, H.; Rasmussen, A.L.; et al. Ebolaviruses associated with differential pathogenicity induce distinct host responses in human macrophages. J. Virol. 2017, 91, e00179-17. [Google Scholar] [CrossRef]
- Escudero-Pérez, B.; Volchkova, V.A.; Dolnik, O.; Lawrence, P.; Volchkov, V.E. Shed GP of Ebola virus triggers immune activation and increased vascular permeability. PLoS Pathog. 2014, 10, e1004509. [Google Scholar] [CrossRef]
- Kreuels, B.; Wichmann, D.; Emmerich, P.; Schmidt-Chanasit, J.; de Heer, G.; Kluge, S.; Sow, A.; Renné, T.; Günther, S.; Lohse, A.W.; et al. A case of severe Ebola virus infection complicated by gram-negative septicemia. N. Engl. J. Med. 2014, 371, 2394–2401. [Google Scholar] [CrossRef] [PubMed]
- Rustagi, A.; Gale, M. Innate antiviral immune signaling, viral evasion and modulation by HIV-1. J. Mol. Biol. 2014, 426, 1161–1177. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, T.S.; Ranganath, N.; Angel, J.B. Impairment of the type I interferon response by HIV-1: Potential targets for HIV eradication. Cytokine Growth Factor Rev. 2017, 37, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; Amarasinghe, G.K.; Basler, C.F. Filovirus pathogenesis and immune evasion: Insights from Ebola virus and Marburg virus. Nat. Rev. Microbiol. 2015, 13, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Pérez, B.; Muñoz-Fontela, C. Role of type I interferons on filovirus pathogenesis. Vaccines 2019, 7, 22. [Google Scholar] [CrossRef]
- Liu, X.; Speranza, E.; Muñoz-Fontela, C.; Haldenby, S.; Rickett, N.Y.; Garcia-Dorival, I.; Fang, Y.; Hall, Y.; Elsa-Gayle, Z.; Lüdtke, A.; et al. Transcriptomic signatures differentiate survival from fatal outcomes in humans infected with Ebola virus. Genome Biol. 2017, 18, 4. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Halfmann, P.J.; Wendler, J.P.; Kyle, J.E.; Burnum-Johnson, K.E.; Peralta, Z.; Maemura, T.; Walters, K.B.; Watanabe, T.; Fukuyama, S.; et al. Multi-platform ’omics analysis of human Ebola virus disease pathogenesis. Cell Host Microbe 2017, 22, 817–829. [Google Scholar] [CrossRef]
- Kerber, R.; Krumkamp, R.; Korva, M.; Rieger, T.; Wurr, S.; Duraffour, S.; Oestereich, L.; Gabriel, M.; Sissoko, D.; Anglaret, X.; et al. Kinetics of soluble mediators of the host response in Ebola virus disease. J. Infect. Dis. 2018, 218 (Suppl. S5), S496–S503. [Google Scholar] [CrossRef]
- Akiyama, H.; Pina Ramirez, N.-G.; Gibson, G.; Kline, C.; Watkins, S.; Ambrose, Z.; Gummuluru, S. Interferon-inducible CD169/Siglec1 attenuates anti-HIV-1 effects of alpha interferon. J. Virol. 2017, 91, e00972-17. [Google Scholar] [CrossRef]
- Zou, Z.; Chastain, A.; Moir, S.; Ford, J.; Trandem, K.; Martinelli, E.; Cicala, C.; Crocker, P.; Arthos, J.; Sun, P.D. Siglecs facilitate HIV-1 infection of macrophages through adhesion with viral sialic acids. PLoS ONE 2011, 6, e24559. [Google Scholar] [CrossRef]
- Ruffin, N.; Gea-Mallorquí, E.; Brouiller, F.; Jouve, M.; Silvin, A.; See, P.; Duterte, C.-A.; Ginhoux, F.; Benaroch, P. Constitutive Siglec-1 expression confers susceptibility to HIV-1 infection of human dendritic cell precursors. Proc. Natl. Acad. Sci. USA 2019, 116, 21685–21693. [Google Scholar] [CrossRef] [PubMed]
- Granelli-Piperno, A.; Moser, B.; Pope, M.; Chen, D.; Wei, Y.; Isdell, F.; O’Doherty, U.; Paxton, W.; Koup, R.; Mosjov, S.; et al. Efficient interaction of HIV-1 with purified dendritic cells via multiple chemokine coreceptors. J. Exp. Med. 1996, 184, 2433–2438. [Google Scholar] [CrossRef] [PubMed]
- Turville, S.G.; Cameron, P.U.; Handley, A.; Lin, G.; Pöhlmann, S.; Doms, R.W.; Cunningham, A.L. Diversity of receptors binding HIV on dendritic cell subsets. Nat. Immunol. 2002, 3, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Knight, S.C. Susceptibility of human peripheral blood dendritic cells to infection by human immunodeficiency virus. J. Gen. Virol. 1987, 68, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Langhoff, E.; Terwilliger, E.F.; Bos, H.J.; Kalland, K.H.; Poznansky, M.C.; Bacon, O.M.L.; Haseltine, W.A. Replication of human immunodeficieacy virus type 1 in primary dendritic cell cultures. Proc. Natl. Acad. Sci. USA 1991, 88, 7998–8002. [Google Scholar] [CrossRef] [PubMed]
- Smed-Sörensen, A.; Lore, K.; Vasudevan, J.; Louder, M.K.; Andersson, J.; Mascola, J.R.; Spetz, A.-L.; Koup, R.A. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J. Virol. 2005, 79, 8861–8869. [Google Scholar] [CrossRef] [PubMed]
- Granelli-Piperno, A.; Delgado, E.; Finkel, V.; Paxton, W.; Steinman, R.M. Immature dendritic cells selectively replicate macrophagetropic (M-tropic) human immunodeficiency virus type 1, while mature cells efficiently transmit both M- and T-tropic virus to T cells. J. Virol. 1998, 72, 2733–2737. [Google Scholar] [PubMed]
- Granelli-Piperno, A.; Finkel, V.; Delgado, E.; Steinman, R.M. Virus replication begins in dendritic cells during the transmission of HIV-1 from mature dendritic cells to T cells. Curr. Biol. 1999, 9, 21–29. [Google Scholar] [CrossRef]
- Cameron, P.U.; Forsum, U.; Teppler, H.; Granelli-Piperno, A.; Steinman, R.M. During HIV-1 infection most blood dendritic cells are not productively infected and can induce allogeneic CD4+ T cells clonal expansion. Clin. Exp. Immunol. 1992, 88, 226–236. [Google Scholar] [CrossRef]
- Pope, M.; Gezelter, S.; Gallo, N.; Hoffman, L.; Steinman, R.M. Low levels of HIV-1 infection in cutaneous dendritic cells promote extensive viral replication upon binding to memory CD4+ T cells. J. Exp. Med. 1995, 182, 2045–2056. [Google Scholar] [CrossRef]
- Bakri, Y.; Schiffer, C.; Zennou, V.; Charneau, P.; Kahn, E.; Benjouad, A.; Gluckman, J.C.; Canque, B. The maturation of dendritic cells results in postintegration inhibition of HIV-1 replication. J. Immunol. 2001, 166, 3780–3788. [Google Scholar] [CrossRef] [PubMed]
- Cavrois, M.; Neidleman, J.; Kreisberg, J.F.; Fenard, D.; Callebaut, C.; Greene, W.C. Human immunodeficiency virus fusion to dendritic cells declines as cells mature. J. Virol. 2006, 80, 1992–1999. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; KewalRamani, V.N. Dendritic-cell interactions with HIV: Infection and viral dissemination. Nat. Rev. Immunol. 2006, 6, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Frank, I.; Williams, V.; Santos, J.J.; Watts, P.; Griffin, G.E.; Moore, J.P.; Pope, M.; Shattock, R.J. Blockade of attachment and fusion receptors inhibits HIV-1 infection of human cervical tissue. J. Exp. Med. 2004, 199, 1065–1075. [Google Scholar] [CrossRef]
- Pena-Cruz, V.; Agosto, L.M.; Akiyama, H.; Olson, A.; Moreau, Y.; Larrieux, J.R.; Henderson, A.; Gummuluru, S.; Sagar, M. HIV-1 replicates and persists in vaginal epithelial dendritic cells. J. Clin. Investig. 2018, 128, 3439–3444. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar] [CrossRef]
- Lahaye, X.; Satoh, T.; Gentili, M.; Cerboni, S.; Conrad, C.; Hurbain, I.; ElMarjou, A.; Lacabaratz, C.; Lelièvre, J.D.; Manel, N. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 2013, 39, 1132–1142. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2014, 341, 903–906. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Buseyne, F.; Le Gall, S.; Boccaccio, C.; Abastado, J.-P.; Lifson, J.D.; Arthur, L.O.; Rivière, Y.; Heard, J.-M.; Schwartz, O. MHC-I–restricted presentation of HIV-1 virion antigens without viral replication. Nat. Med. 2001, 7, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Bosio, C.M.; Aman, M.J.; Grogan, C.; Hogan, R.; Ruthel, G.; Negley, D.; Mohamadzadeh, M.; Bavari, S.; Schmaljohn, A. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J. Infect. Dis. 2003, 188, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Shtanko, O.; Anantpadma, M.; Sakurai, Y.; Chandran, K.; Mauri, W. Mechanisms of filovirus entry. Curr. Top. Microbiol. Immunol. 2017, 411, 323–352. [Google Scholar] [PubMed]
- Zapatero-Belinchón, F.J.; Dietzel, E.; Dolnik, O.; Döhner, K.; Costa, R.; Hertel, B.; Veselkova, B.; Kirui, J.; Klintworth, A.; Manns, M.P.; et al. Characterization of the filovirus-resistant cell line SH-SY5Y reveals redundant role of cell surface entry factors. Viruses 2019, 11, 275. [Google Scholar] [CrossRef]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muñiz, O.; Corbí, A.L.; Delgado, R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar] [CrossRef]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468–470, 565–580. [Google Scholar] [CrossRef]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science (80-) 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Ebola virus entry: A curious and complex series of events. PLoS Pathog. 2015, 11, e1004731. [Google Scholar] [CrossRef]
- Rasmussen, A.L. Host factors in Ebola infection. Annu. Rev. Genomics Hum. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.L.; Lennemann, N.J.; Maury, W. Filovirus entry: A novelty in the viral fusion world. Viruses 2012, 4, 258–275. [Google Scholar] [CrossRef]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; Mcrae, M.; Rollin, P.E.; Pulendran, B. Impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar] [CrossRef]
- Bray, M.; Geisbert, T.W. Ebola virus: The role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 2005, 37, 1560–1566. [Google Scholar] [CrossRef]
- Martinez, O.; Leung, L.W.; Basler, C.F. The role of antigen-presenting cells in filoviral hemorrhagic fever: Gaps in current knowledge. Antivir. Res. 2012, 93, 416–428. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.K.; Akondy, R.S.; Davis, C.W.; Ellebedy, A.H.; Mehta, A.K.; Kraft, C.S.; Lyon, G.M.; Ribner, B.S.; Varkey, J.; Sidney, J.; et al. Human Ebola virus infection results in substantial immune activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4719–4724. [Google Scholar] [CrossRef]
- Silvin, A.; Yu, C.I.; Lahaye, X.; Imperatore, F.; Brault, J.B.; Cardinaud, S.; Becker, C.; Kwan, W.-H.; Conrad, C.; Maurin, M.; et al. Constitutive resistance to viral infection in human CD141+ dendritic cells. Sci. Immunol. 2017, 2, eaai8071. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Théry, C.; Duban, L.; Segura, E.; Véron, P.; Lantz, O.; Amigorena, S. Indirect activation of naïve CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Useros, N.; Naranjo-Gómez, M.; Archer, J.; Hatch, S.C.; Erkizia, I.; Blanco, J.; Borràs, F.E.; Puertas, M.C.; Connor, J.H.; Fernández-Figueras, M.T.; et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 2009, 113, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.M.; Fang, Y.; Fallon, J.K.; Yang, J.M.; Hildreth, J.E.K.; Gould, S.J. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J. Cell Biol. 2006, 172, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wu, N.; Gan, X.; Yan, W.; Morrell, J.C.; Gould, S.J. Higher-order oligomerization targets plasma membrane proteins and HIV Gag to exosomes. PLoS Biol. 2007, 5, e158. [Google Scholar] [CrossRef]
- Díaz-Varela, M.; de Menezes-Neto, A.; Perez-Zsolt, D.; Gámez-Valero, A.; Seguí-Barber, J.; Izquierdo-Useros, N.; Martinez-Picado, J.; Fernández-Becerra, C.; del Portillo, H.A. Proteomics study of human cord blood reticulocyte-derived exosomes. Sci. Rep. 2018, 8, 14046. [Google Scholar] [CrossRef]
- Saunderson, S.C.; Dunn, A.C.; Crocker, P.R.; McLellan, A.D. CD169 mediates the capture of exosomes in spleen and lymph node. Blood 2014, 123, 208–216. [Google Scholar] [CrossRef]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef]
- Platt, C.D.; Ma, J.K.; Chalouni, C.; Ebersold, M.; Bou-Reslan, H.; Carano, R.A.D.; Mellman, I.; Delamarre, L. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc. Natl. Acad. Sci. USA 2010, 107, 4287–4292. [Google Scholar] [CrossRef]
- Drutman, S.B.; Trombetta, E.S. Dendritic cells continue to capture and present antigens after maturation in vivo. J. Immunol. 2010, 185, 2140–2146. [Google Scholar] [CrossRef]
- McDonald, D.; Wu, L.; Bohks, S.M.; KewalRamani, V.N.; Unutmaz, D.; Hope, T.J. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science (80-) 2003, 300, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Plata, M.T.; Puigdomènech, I.; Izquierdo-Useros, N.; Puertas, M.C.; Carrillo, J.; Erkizia, I.; Clotet, B.; Blanco, J.; Martinez-Picado, J. The infectious synapse formed between mature dendritic cells and CD4+ T cells is independent of the presence of the HIV-1 envelope glycoprotein. Retrovirology 2013, 10, 42. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.H.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.F.; Middel, J.; Cornelissen, I.L.M.H.A.; Nottet, H.S.L.M.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Gibb, T.R.; Steele, K.E.; Jaax, N.K.; Jahrling, P.B. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Investig. 2000, 80, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Baskerville, A.; Fisher-Hoch, S.P.; Neild, G.H.; Dowsett, A.B. Ultrastructural pathology of experimental Ebola haemorrhagic fever virus infection. J. Pathol. 1985, 147, 199–209. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jahrling, P.B.; Hanes, M.A.; Zack, P.M. Association of Ebola-related Reston virus particles and antigen with tissue lesions of monkeys imported to the United States. J. Comp. Pathol. 1992, 106, 137–152. [Google Scholar] [CrossRef]
- Davis, K.J.; Anderson, A.O.; Geisbert, T.W.; Steele, K.E.; Geisbert, J.B.; Vogel, P.; Connolly, B.M.; Huggins, J.W.; Jahrling, P.B.; Jaax, N.K. Pathology of experimental Ebola virus infection in African green monkeys: Involvement of fibroblastic reticular cells. Arch. Pathol. Lab. Med. 1997, 121, 805–819. [Google Scholar]
- Geisbert, T.W.; Jaax, N.K. Marburg hemorrhagic fever: Report of a case studied by immunohistochemistry and electron microscopy. Ultrastruct. Pathol. 1998, 22, 3–17. [Google Scholar] [CrossRef]
- Lasala, F.; Arce, E.; Otero, J.R.; Rojo, J.; Delgado, R. Mannosyl glycodendritic structure inhibits DC-SIGN-mediated Ebola virus infection in cis and in trans. Antimicrob. Agents Chemother. 2003, 47, 3970–3972. [Google Scholar] [CrossRef]
- Van Kooyk, Y.; Geijtenbeek, T.B.H. DC-SIGN: Escape mechanism for pathogens. Nat. Rev. Immunol. 2003, 3, 697–709. [Google Scholar] [CrossRef]
- Kader, M.; Smith, A.P.; Guiducci, C.; Wonderlich, E.R.; Normolle, D.; Watkins, S.C.; Barrat, F.J.; Barratt-Boyes, S.M. Blocking TLR7- and TLR9-mediated IFN-α production by plasmacytoid dendritic cells does not diminish immune activation in early SIV infection. PLoS Pathog. 2013, 9, e1003530. [Google Scholar] [CrossRef]
- Li, Q.; Estes, J.D.; Schlievert, P.M.; Duan, L.; Brosnahan, A.J.; Southern, P.J.; Reilly, C.S.; Peterson, M.L.; Schultz-Darken, N.; Brunner, K.G.; et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature 2009, 458, 1034–1038. [Google Scholar] [CrossRef]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef]
- Masurier, C.; Salomon, B.; Guettari, N.; Pioche, C.; Lachapelle, F.; Guigon, M.; Klatzmann, D. Dendritic cells route human immunodeficiency virus to lymph nodes after vaginal or intravenous administration to mice. J. Virol. 1998, 72, 7822–7829. [Google Scholar]
- Hu, J.; Gardner, M.B.; Miller, C.J. Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J. Virol. 2000, 74, 6087–6095. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Li, Q.; Abel, K.; Kim, E.-Y.; Ma, Z.-M.; Wietgrefe, S.; La Franco-Scheuch, L.; Compton, L.; Duan, L.; Shore, M.D.; et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J. Virol. 2005, 79, 9217–9227. [Google Scholar] [CrossRef]
- Mate, S.E.; Kugelman, J.R.; Nyenswah, T.G.; Ladner, J.T.; Wiley, M.R.; Cordier-Lassalle, T.; Christie, A.; Schroth, G.P.; Gross, S.M.; Davies-Wayne, G.J.; et al. Molecular evidence of sexual transmission of Ebola virus. N. Engl. J. Med. 2015, 373, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Zheng, R.; Wang, D.; Zhang, X.; Yin, Y.; Wang, K.; Wang, W. Effect of sexual transmission on the West Africa Ebola outbreak in 2014: A mathematical modelling study. Sci. Rep. 2019, 9, 1653. [Google Scholar] [CrossRef]
- Emond, R.T.D.; Evans, B.; Bowen, E.T.W.; Lloyd, G. A case of Ebola virus infection. Br. Med. J. 1977, 2, 541–544. [Google Scholar] [CrossRef]
- Bausch, D.G.; Towner, J.S.; Dowell, S.F.; Kaducu, F.; Lukwiya, M.; Sanchez, A.; Nichol, S.T.; Ksiazek, T.G.; Rollin, P.E. Assessment of the risk of Ebola virus Transmission from bodily fluids and fomites. J. Infect. Dis. 2007, 196 (Suppl. S2), S142–S147. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.L.; De Roo, A.; Guimard, Y.; Trappier, S.G.; Sanchez, A.; Bressler, D.; Williams, A.J.; Rowe, A.K.; Bertolli, J.; Khan, A.S.; et al. Persistence and genetic stability of Ebola virus during the outbreak in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999, 179 (Suppl. S1), S170–S176. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.K.; Bertolli, J.; Khan, A.S.; Mukunu, R.; Muyembe-Tamfum, J.J.; Bressler, D.; Williams, A.J.; Peters, C.J.; Rodriguez, L.; Feldmann, H.; et al. Clinical, virologic, and immunologic follow-up of convalescent Ebola hemorrhagic fever patients and their household contacts, Kikwit, Democratic Republic of the Congo. J. Infect. Dis. 1999, 179 (Suppl. S1), S28–S35. [Google Scholar] [CrossRef] [PubMed]
- Bart, S.M.; Cohen, C.; Dye, J.M.; Shorter, J.; Bates, P. Enhancement of Ebola virus infection by seminal amyloid fibrils. Proc. Natl. Acad. Sci. USA 2018, 115, 7410–7415. [Google Scholar] [CrossRef] [PubMed]
- De Saint Jean, A.; Lucht, F.; Bourlet, T.; Delézay, O. Transforming growth factor beta 1 up-regulates CD169 (sialoadhesin) expression on monocytederived dendritic cells: Role in HIV sexual transmission. Aids 2014, 28, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Steiniger, B.; Barth, P.; Herbst, B.; Hartnell, A.; Crocker, P.R. The species-specific structure of microanatomical compartments in the human spleen: Strongly sialoadhesin-positive macrophages occur in the perifollicular zone, but not in the marginal zone. Immunology 1997, 92, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999, 179 (Suppl. S1), S199–S202. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F. Molecular pathogenesis of viral hemorrhagic fever. Semin. Immunopathol. 2017, 39, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Miller, C.; Patel, H.V.; Hatch, S.C.; Archer, J.; Ramirez, N.-G.P.; Gummuluru, S. Virus particle release from glycosphingolipid-enriched microdomains is essential for dendritic cell-mediated capture and transfer of HIV-1 and henipavirus. J. Virol. 2014, 88, 8813–8825. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; McLaren, P.J.; Erkizia, I.; Martin, M.P.; Benet, S.; Rotger, M.; Dalmau, J.; Ouchi, D.; Wolinsky, S.M.; Penugonda, S.; et al. Identification of Siglec-1 null individuals infected with HIV-1. Nat. Commun. 2016, 7, 12412. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Zsolt, D.; Martinez-Picado, J.; Izquierdo-Useros, N. When Dendritic Cells Go Viral: The Role of Siglec-1 in Host Defense and Dissemination of Enveloped Viruses. Viruses 2020, 12, 8. https://doi.org/10.3390/v12010008
Perez-Zsolt D, Martinez-Picado J, Izquierdo-Useros N. When Dendritic Cells Go Viral: The Role of Siglec-1 in Host Defense and Dissemination of Enveloped Viruses. Viruses. 2020; 12(1):8. https://doi.org/10.3390/v12010008
Chicago/Turabian StylePerez-Zsolt, Daniel, Javier Martinez-Picado, and Nuria Izquierdo-Useros. 2020. "When Dendritic Cells Go Viral: The Role of Siglec-1 in Host Defense and Dissemination of Enveloped Viruses" Viruses 12, no. 1: 8. https://doi.org/10.3390/v12010008
APA StylePerez-Zsolt, D., Martinez-Picado, J., & Izquierdo-Useros, N. (2020). When Dendritic Cells Go Viral: The Role of Siglec-1 in Host Defense and Dissemination of Enveloped Viruses. Viruses, 12(1), 8. https://doi.org/10.3390/v12010008