RNase L Is Involved in Liposaccharide-Induced Lung Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Knocking Down of RNase L in Raw 264.7 Cells
2.3. Animal Treatment
2.4. Analysis of RNase L-Mediated rRNA Cleavages in Intact Cells
2.5. Western Blot Analysis
2.6. Histostaining
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
3. Results
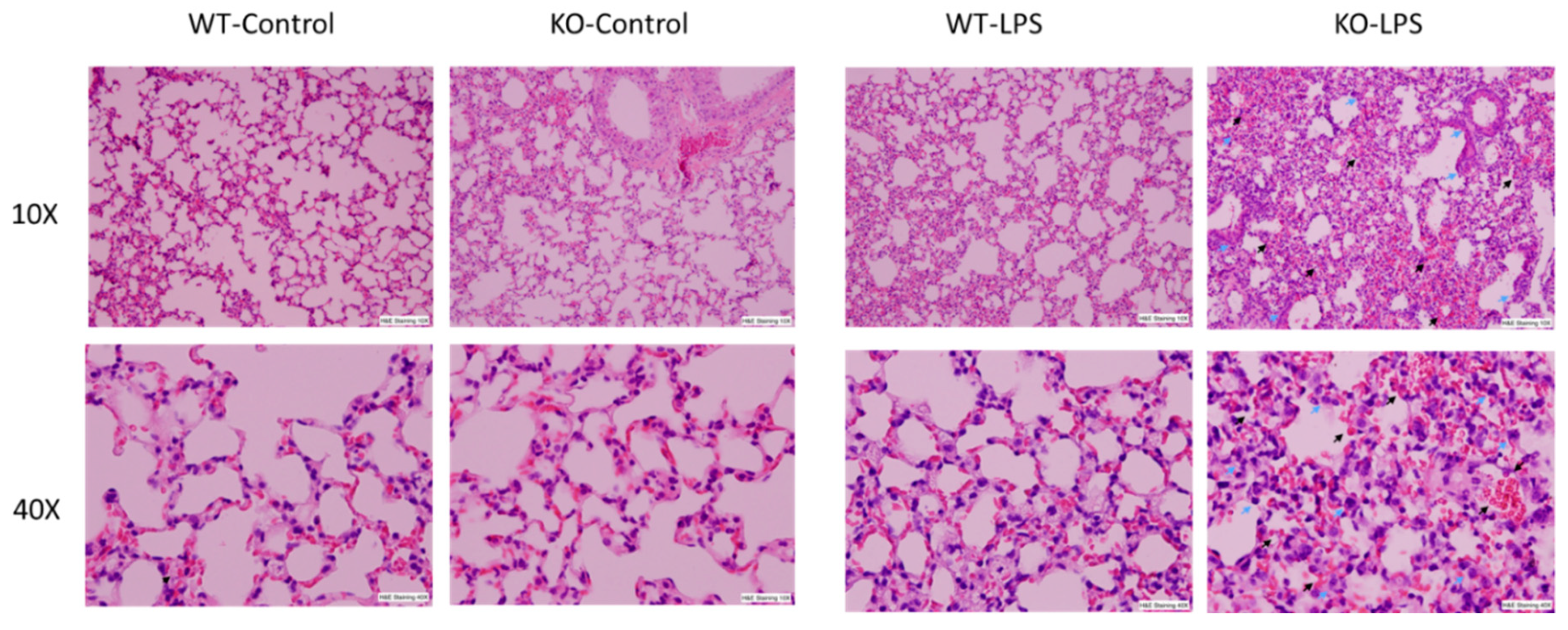
3.1. Deficiency of RNase L Increases Lung Tissue Damage in LPS-Induced ALI
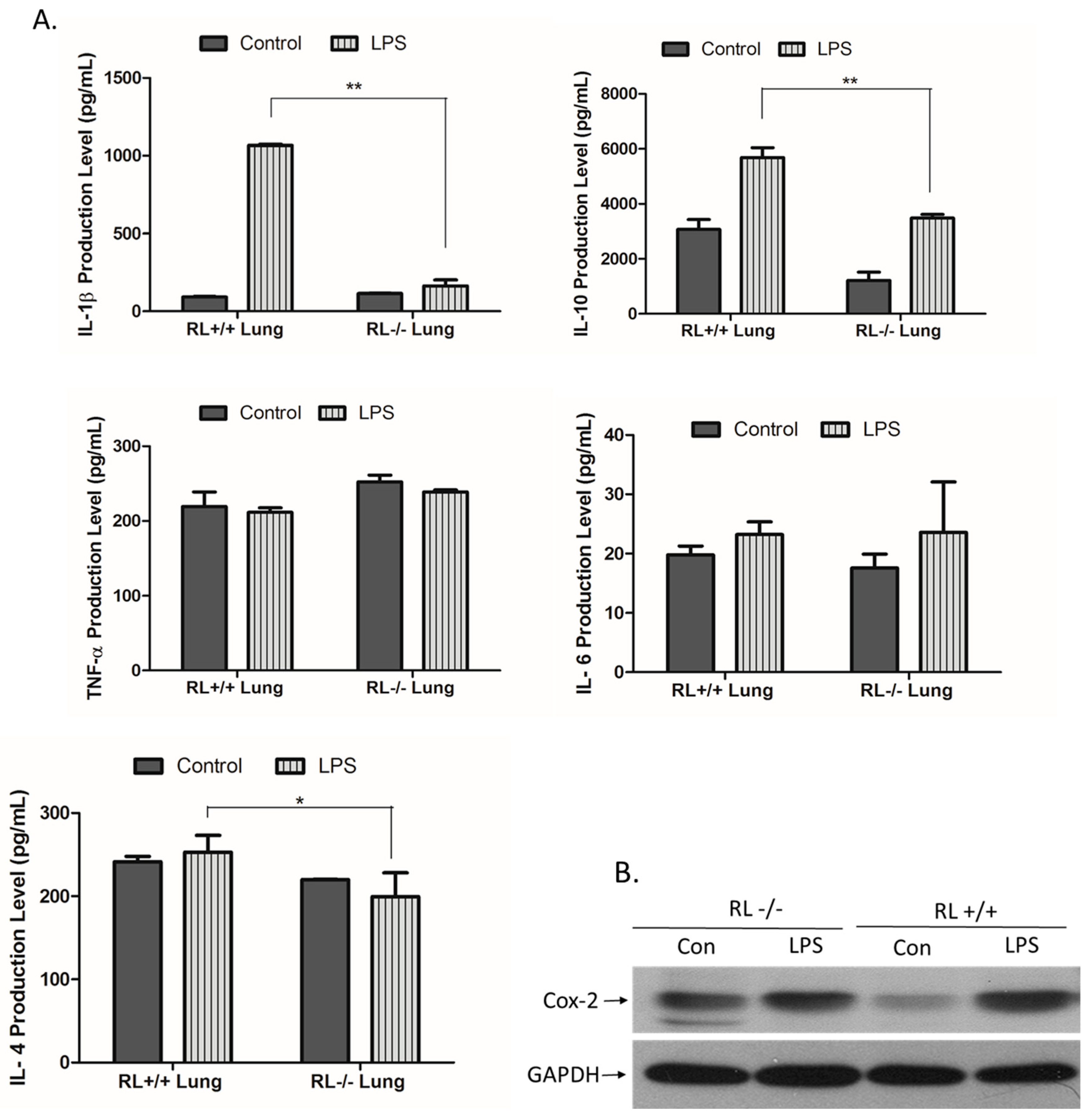
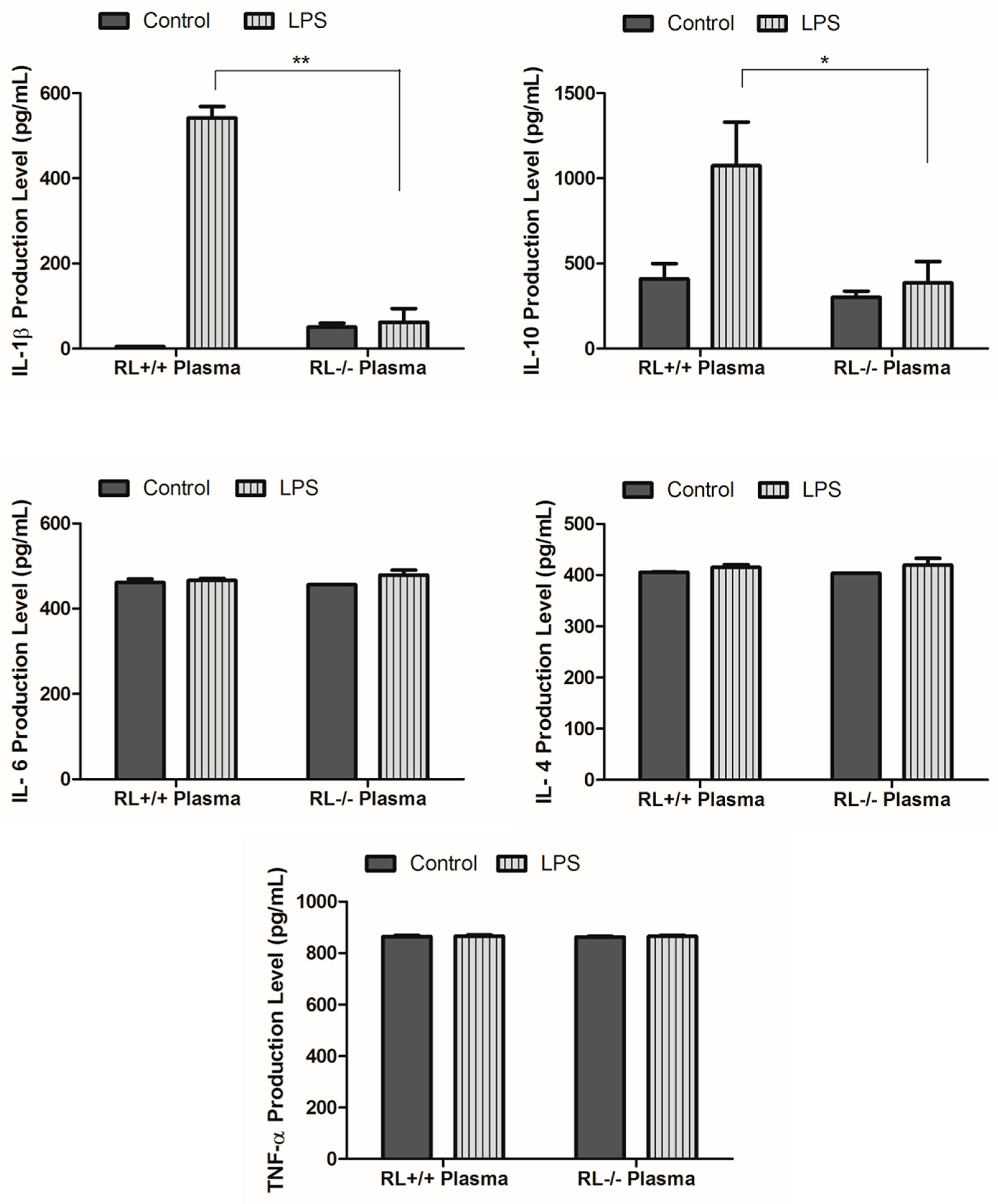
3.2. RNase L Regulates the Expression of Pro-and Anti-Inflammatory Genes in the Lung and Blood upon LPS Stimulation
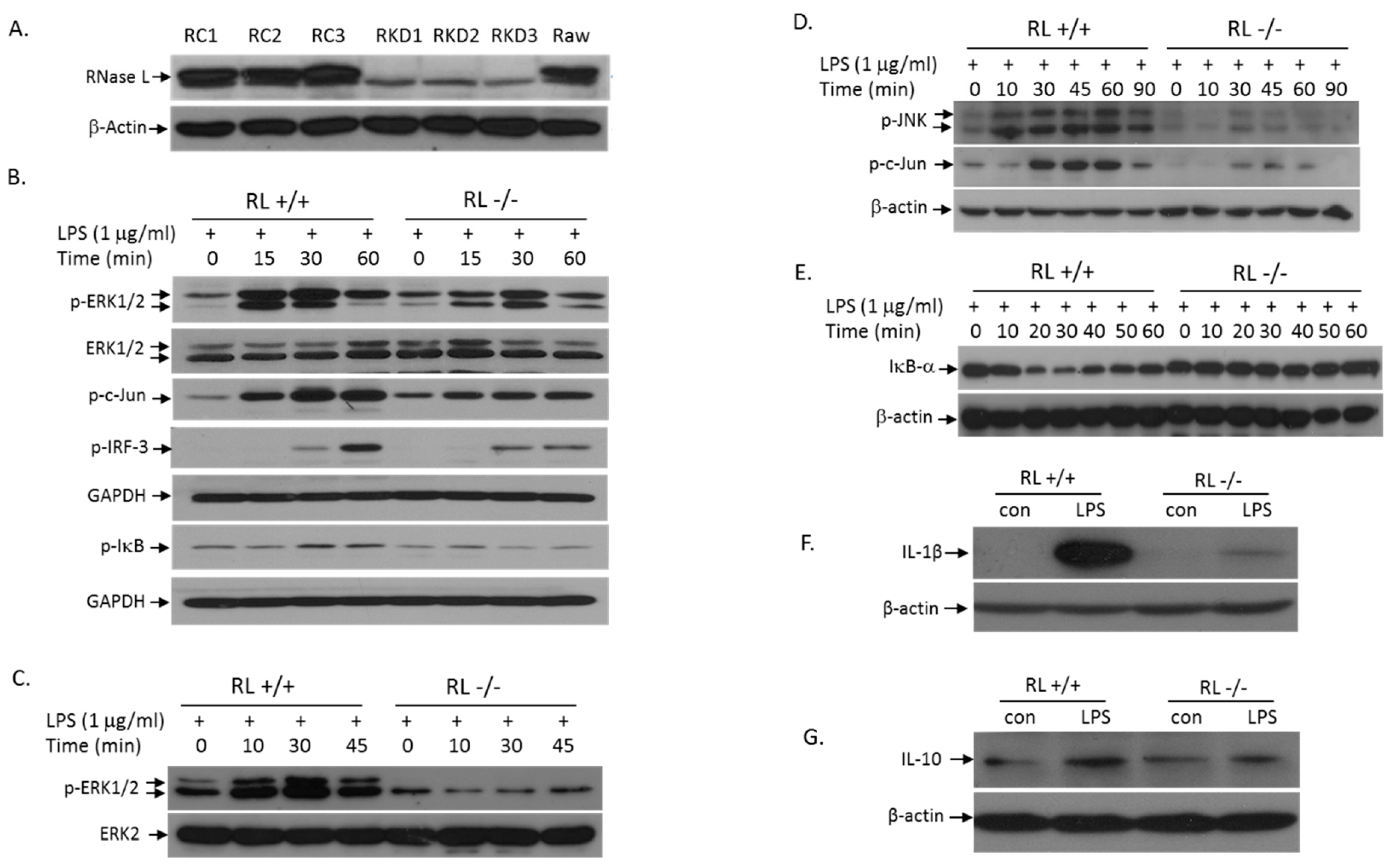
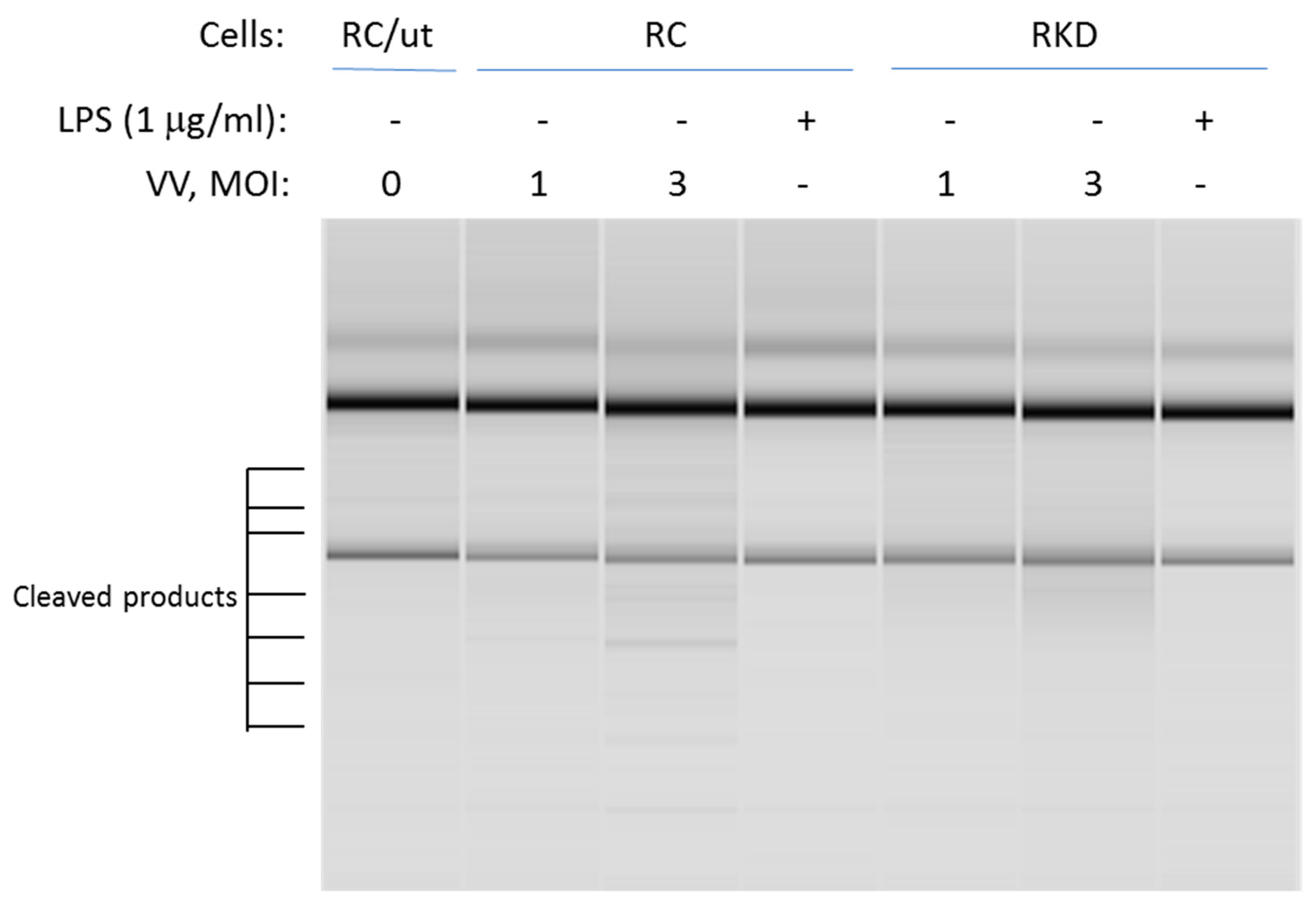
3.3. RNase L Mediates the TLR4 Signaling Pathway Activated by LPS in Macrophages
3.4. RNase L Function in LPS-Induced Lung Injury May Be Independent of Its Nuclease Activity
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef]
- Wheeler, A.P.; Bernard, G.R. Acute lung injury and the acute respiratory distress syndrome: A clinical review. Lancet 2007, 369, 1553–1564. [Google Scholar] [CrossRef]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef]
- Lomas-Neira, J.; Chung, C.S.; Perl, M.; Gregory, S.; Biffl, W.; Ayala, A. Role of alveolar macrophage and migrating neutrophils in hemorrhage-induced priming for ALI subsequent to septic challenge. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L51–L58. [Google Scholar] [CrossRef]
- Johnston, L.K.; Rims, C.R.; Gill, S.E.; McGuire, J.K.; Manicone, A.M. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 417–426. [Google Scholar] [CrossRef]
- Silverman, R.H. 16—2-5A-Dependent RNase L: A regulated endoribonuclease in the interferon system. In Ribonucleases; D’Alessio, G., Riordan, J.F., Eds.; Academic Press: New York, NY, USA, 1997; pp. 515–551. [Google Scholar] [CrossRef]
- Zhou, A.; Hassel, B.A.; Silverman, R.H. Expression cloning of 2-5A-dependent RNAase: A uniquely regulated mediator of interferon action. Cell 1993, 72, 753–765. [Google Scholar] [CrossRef]
- Zhou, A.; Paranjape, J.M.; Hassel, B.A.; Nie, H.; Shah, S.; Galinski, B.; Silverman, R.H. Impact of RNase L overexpression on viral and cellular growth and death. J. Interferon Cytokine Res. 1998, 18, 953–961. [Google Scholar] [CrossRef]
- Hassel, B.A.; Zhou, A.; Sotomayor, C.; Maran, A.; Silverman, R.H. A dominant negative mutant of 2-5A-dependent RNase suppresses antiproliferative and antiviral effects of interferon. EMBO J. 1993, 12, 3297–3304. [Google Scholar] [CrossRef]
- Zhou, A.; Paranjape, J.; Brown, T.L.; Nie, H.; Naik, S.; Dong, B.; Chang, A.; Trapp, B.; Fairchild, R.; Colmenares, C.; et al. Interferon action and apoptosis are defective in mice devoid of 2’,5’-oligoadenylate-dependent RNase L. EMBO J. 1997, 16, 6355–6363. [Google Scholar] [CrossRef]
- Silverman, R.H. Viral encounters with 2’,5’-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef]
- Rusch, L.; Zhou, A.; Silverman, R.H. Caspase-dependent apoptosis by 2’,5’-oligoadenylate activation of RNase L is enhanced by IFN-beta. J. Interferon Cytokine Res. 2000, 20, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Castelli, J.C.; Hassel, B.A.; Maran, A.; Paranjape, J.; Hewitt, J.A.; Li, X.L.; Hsu, Y.T.; Silverman, R.H.; Youle, R.J. The role of 2’-5’ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998, 5, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Malathi, K. RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 2012, 287, 43651–43664. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Banerjee, S.; Franchi, L.; Loo, Y.-M.; Gale, M.; Núñez, G.; Silverman, R.H. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 2015, 17, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yi, X.; Zipris, D.; Liu, H.; Zhang, L.; Zheng, Q.; Malathi, K.; Jin, G.; Zhou, A. RNase L contributes to experimentally induced type 1 diabetes onset in mice. J. Endocrinol. 2014, 223, 277–287. [Google Scholar] [CrossRef]
- Silverman, R.H.; Zhou, A.; Auerbach, M.B.; Kish, D.; Gorbachev, A.; Fairchild, R.L. Skin allograft rejection is suppressed in mice lacking the antiviral enzyme, 2’,5’-oligoadenylate-dependent RNase L. Viral Immunol. 2002, 15, 77–83. [Google Scholar] [CrossRef]
- Leitner, W.W.; Hwang, L.N.; deVeer, M.J.; Zhou, A.; Silverman, R.H.; Williams, B.R.; Dubensky, T.W.; Ying, H.; Restifo, N.P. Alphavirus-based DNA vaccine breaks immunological tolerance by activating innate antiviral pathways. Nat. Med. 2003, 9, 33–39. [Google Scholar] [CrossRef]
- Yi, X.; Zeng, C.; Liu, H.; Chen, X.; Zhang, P.; Yun, B.S.; Jin, G.; Zhou, A. Lack of RNase L attenuates macrophage functions. PLoS ONE 2013, 8, e81269. [Google Scholar] [CrossRef]
- Martin, W.J., II; Wu, M.; Pasula, R. A novel approach to restore lung immunity during systemic immunosuppression. Trans. Am. Clin. Clim. Assoc. 2005, 116, 221–226. [Google Scholar]
- Kapur, R.; Kim, M.; Aslam, R.; McVey, M.J.; Tabuchi, A.; Luo, A.; Liu, J.; Li, Y.; Shanmugabhavananthan, S.; Speck, E.R.; et al. T regulatory cells and dendritic cells protect against transfusion-related acute lung injury via IL-10. Blood 2017, 129, 2557–2569. [Google Scholar] [CrossRef]
- Inoue, G. Effect of interleukin-10 (IL-10) on experimental LPS-induced acute lung injury. J. Infect. Chemother. 2000, 6, 51–60. [Google Scholar] [CrossRef]
- Kudo, D.; Toyama, M.; Aoyagi, T.; Akahori, Y.; Yamamoto, H.; Ishii, K.; Kanno, E.; Maruyama, R.; Kaku, M.; Kushimoto, S.; et al. Involvement of high mobility group box 1 and the therapeutic effect of recombinant thrombomodulin in a mouse model of severe acute respiratory distress syndrome. Clin. Exp. Immunol. 2013, 173, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Cochet, F.; Peri, F. The Role of Carbohydrates in the Lipopolysaccharide (LPS)/Toll-Like Receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 2017, 18, 2318. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, R.; Chen, G.; Algehainy, N.; Zeng, C.; Liu, C.; Liu, H.; Liu, W.; Stacey, D.; Zhou, A. RNase L Is Involved in Liposaccharide-Induced Lung Inflammation. Viruses 2020, 12, 73. https://doi.org/10.3390/v12010073
Wei R, Chen G, Algehainy N, Zeng C, Liu C, Liu H, Liu W, Stacey D, Zhou A. RNase L Is Involved in Liposaccharide-Induced Lung Inflammation. Viruses. 2020; 12(1):73. https://doi.org/10.3390/v12010073
Chicago/Turabian StyleWei, Ruhan, Guanmin Chen, Naseh Algehainy, Chun Zeng, Chunfang Liu, Hongli Liu, Wendy Liu, Dennis Stacey, and Aimin Zhou. 2020. "RNase L Is Involved in Liposaccharide-Induced Lung Inflammation" Viruses 12, no. 1: 73. https://doi.org/10.3390/v12010073
APA StyleWei, R., Chen, G., Algehainy, N., Zeng, C., Liu, C., Liu, H., Liu, W., Stacey, D., & Zhou, A. (2020). RNase L Is Involved in Liposaccharide-Induced Lung Inflammation. Viruses, 12(1), 73. https://doi.org/10.3390/v12010073